В настоящем разделе мы попытаемся вывести математические выражения, характеризующие скорости катализируемых ферментами реакций. Естественно, важнейшим критерием правильности такого выражения будет соответствие вычисленных и экспериментально найденных скоростей. Чтобы исключить возможные недоразумения, сначала мы в общих чертах опишем некоторые экспериментальные методы определения скоростей реакций.
Прежде всего определим, что мы имеем в виду под скоростью реакции. Рассмотрим реакцию
![]()
Скорость этой реакции в приближении квазиравновесного состояния (раздел 3.2.1) определяется как
![]()
где символами s и р обозначены молярные концентрации исходного вещества S и продукта реакции Р соответственно. Отсюда следует, что v выражается в числе молей в единице объема в единицу времени. Скорость реакции является интенсивной величиной, имеющей определенное значение в каждой точке реакционной смеси. Поэтому, если концентрации или другие интенсивные переменные изменяются от точки к точке, то и скорости реакции в этих точках будут различными. При экспериментальном изучении кинетики часто используют реакторы с эффективным перемешиванием, в которых обеспечивается одна и та же скорость реакции во всем объеме реакционной смеси.
Как и при моделировании любых других технологических процессов, слово «точка» в определении скорости реакции используется не в строго геометрическом смысле. Напротив, здесь под точкой подразумевается некоторый объем, в который входит множество молекул, но который тем не менее очень мал по сравнению с объемом всей реакционной смеси. Это на первый взгляд не столь существенное уточнение очень важно не забывать при переходе от гипотетических моделей к конкретным биологическим системам. Например, при моделировании молекулярных процессов в отдельной изолированной клетке понятия о концентрации данного вещества Al и скорости его превращения в определенной точке могут оказаться вообще неприменимыми, если в клетке содержится только небольшое число молекул Аl.
Типичный эксперимент по изучению кинетики ферментативных реакций (т. е. реакций, катализируемых ферментами) обычно проводят следующим образом. В нулевой момент времени растворы субстрата и соответствующего очищенного фермента смешивают при эффективном перемешивании в закрытом сосуде, в котором поддерживается постоянная температура и который содержит буферный раствор, необходимый для поддержания определенного значения рН. Далее через заданные промежутки времени определяют концентрации субстрата и (или) продукта реакции. Для этой цели применяют различные методы, в том числе спектрофотометрические, манометрические, электродные, поляриметрические, а также методы, связанные с отбором проб. Обычно используют только данные о начальных скоростях реакций. Поскольку условия реакции, в том числе концентрации фермента и субстрата, точнее всего известны при нулевом времени реакции, начальный наклон кривой, отражающей изменение концентрации субстрата или продукта реакции в- зависимости от времени, определяется по следующим данным:
![]()
На рис. 3.5 приведены данные, полученные в одном из экспериментов описанного типа. Обратите внимание на то, что при t=0 в смеси уже имеется некоторое количество продукта реакции; отсюда следует, что нулевое время на самом деле не является временем начала реакции. В этом заключается одна из трудностей, присущих методу определения начальных скоростей реакций; другие недостатки метода описаны в специальной литературе по химической кинетике. Тем не менее метод начальных скоростей позволяет с достаточной воспроизводимостью определять ферментативную активность и концентрации веществ в первый момент реакции и поэтому с успехом может применяться в практической работе.
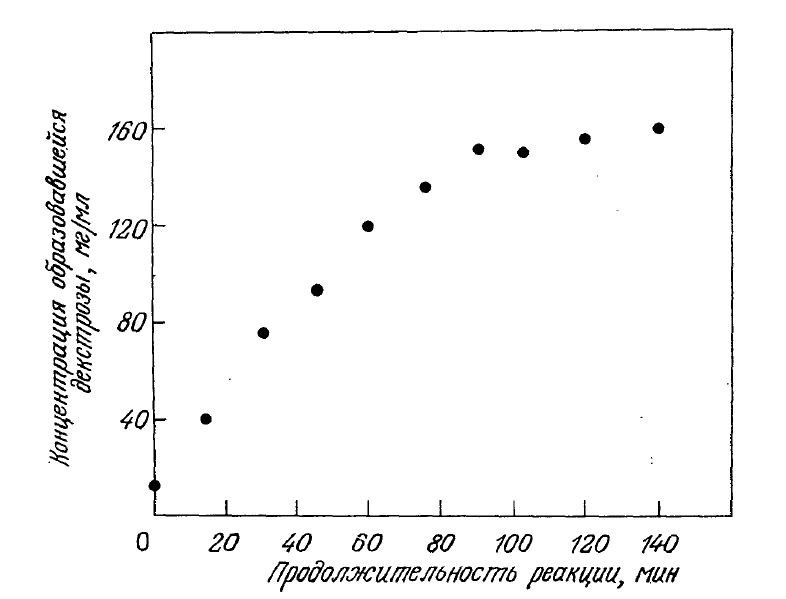
РИС. 3.5. Экспериментальное определение скорости гидролиза 30%-го раствора крахмала
глюкоамилазой в реакторе периодического действия (60°С, е0=11 600 единиц, объем реактора 1л).
Проблема воспроизводимости результатов определения ферментативной активности важна со многих точек зрения, в том числе и с точки зрения конструкций реакторов, в которых осуществляются процессы с изолированными ферментами. В предыдущих разделах мы уже упоминали, что белки в условиях, отличающихся от свойственного им биологического окружения, легко подвергаются денатурации; в этой связи неудивительно, что изолированный фермент в «необычной» для него водной среде может постепенно терять свою каталитическую активность (рис. 3.6). Известно, что такая инактивация характерна для многих ферментов; в то же время во многих руководствах по кинетике ферментативного катализа это явление в лучшем случае только упоминается. Проблема постепенной потери каталитической активности ферментами не столь существенна in vivo (в интактном живом организме), когда снижение концентраций ферментов за счет инактивации компенсируется их биосинтезом в необходимых количествах. Напротив, инактивацию фермента in vitro (вне живой клетки) необходимо учитывать как при изучении кинетики реакций, так и при проектировании ферментативных реакторов. К этой теме мы обратимся еще раз в разд. 3.7.
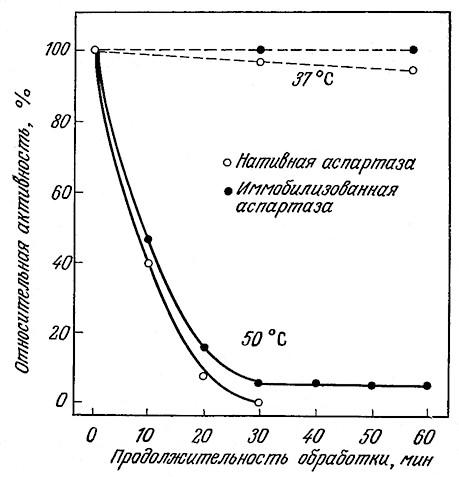
РИС. 3.6. Инактивация фермента в растворе.
В течение 1 ч при 37 °С раствор аспартазы теряет более 5% исходной активности.
В подписи к рис. 3.5 (в скобках) нам впервые встречается другая типичная для кинетики ферментативного катализа особенность. Обратите внимание на то, что количество фермента, использовавшегося в этом эксперименте, выражено в неких «единицах». Что это за таинственные единицы и почему нельзя количество фермента выразить более понятным и более определенным способом, например, в молях или в единицах массы?
Сначала ответим на вторую часть вопроса. Количество фермента обычно не выражают, например, в единицах массы, поскольку ферментные препараты, как правило, представляют собой смесь различных белков, в которой данный фермент является всего лишь одним из нескольких компонентов. Содержание интересующего нас фермента в этой смеси часто неизвестно; более того, оно может быть различным в разных партиях препарата. Чтобы охарактеризовать препарат, содержание фермента обычно выражают в единицах каталитической активности, содержащейся в единице массы ферментного препарата.
Единицей или единицей активности называют количество фермента, которое обладает определенной каталитической активностью в заданных для этого фермента стандартных условиях. Например, в эксперименте по гидролизу крахмала, результаты которого приведены на рис. 3.5, за единицу активности глюкоамилазы принимали количество фермента, которое в 4%-ном растворе крахмала Линтнера при рН 4,5 и температуре 60 °С образует 1 мкмоль глюкозы в 1 мин. Отсюда следует, что единица активности каждого конкретного фермента будет иметь свое специфическое определение и, более того, один фермент может иметь несколько определений активности в зависимости от конкретных условий катализируемых им реакций., Поэтому во избежание ошибок при интерпретации или использовании результатов изучения кинетики реакций ферментативного катализа всегда следует тщательно проверять применяемое в каждом случае определение единицы ферментативной активности. Конечно, вероятность ошибки резко снижается, если известна активность высокоочищенного фермента.
3.2.1. Уравнение Михаэлиса – Ментен
Предположим теперь, что в нашем распоряжении имеется целый ряд экспериментальных данных по определению скорости ферментативной реакции (рис. 3.7) и перед нами стоит задача выразить эти данные в математической форме. Информация типа показанной на рис. 3.7 часто позволяет сделать следующие выводы качественного характера:
1. При относительно низких концентрациях субстрата реакция имеет первый порядок по субстрату.
2. По мере повышения концентрации субстрата порядок реакции по субстрату постепенно снижается от единицы до нуля.
3. Скорость реакции пропорциональна общему количеству фермента в реакционной смеси.
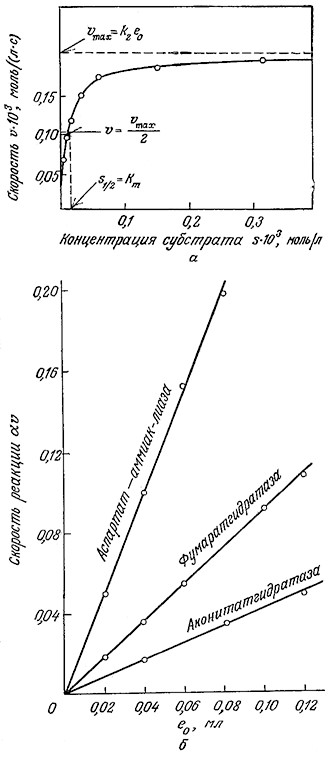
РИС. 3.7. Экспериментальное изучение кинетики катализируемых ферментами реакций.
а – зависимость скорости реакции от концентрации субстрата при постоянной концентрации фермента.
б–зависимость скорости реакции (изменение оптической плотности при 200 нм
за 1 мин) от концентрации фермента при постоянной концентрации субстрата.
На базе экспериментальных данных такого рода в 1902 г. Анри предложил следующее выражение для скорости ферментативной реакции:
![]()
В уравнении (3.3) учтены все три указанные выше закономерности. Обратите внимание, что при s, равном Km, V= Vmax/2. Во избежание недоразумений, связанных с употреблением в литературе различных символов и сокращений, подчеркнем, что s обозначает концентрацию свободного субстрата в реакционной смеси, а е0-–общую концентрацию фермента как в свободном, так и в связанном виде.
Хотя Анри дал теоретическое толкование уравнению (3.3) на базе гипотетического механизма реакции, его доказательство, равно как и близкое обоснование, предложенное в 1913 г. Михаэлисом и Ментен, в настоящее время считается в общем случае не строгим. В то же время в этой главе мы неоднократно и не без успеха будем применять общий ход рассуждений Михаэлиса и Ментен (так и не нашедший до сих пор строгого подтверждения) для разработки более сложных моделей кинетики ферментативного катализа. Поэтому сначала мы рассмотрим основные положения гипотезы Михаэлиса – Ментен.
В качестве одного из исходных положений принимают, что фермент Е и субстрат S, вступая в реакцию, образуют комплекс ES, который затем, диссоциирует на продукт реакции Р и свободный (несвязанный) фермент Е:
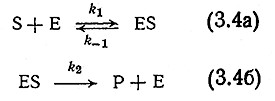
В этом механизме учтены как рассмотренное выше образование фермент-субстратного комплекса, так и регенерация катализатора в исходной форме по завершении последовательности реакций. Уравнения (3.4) в принципе верны, хотя и чрезмерно упрощены.
Анри, а также Михаэлис и Ментен предположили, что реакция (3.4а) обратима, и, следовательно, в соответствии с законом действующих масс мы можем записать:
![]()
Здесь s, е и (es) обозначают концентрации S, Е и ES соответственно. Кроме того, принимается, что процесс разложения комплекса на продукт реакции и свободный фермент необратим:
![]()
Поскольку весь фермент находится или в свободной, или в связанной форме, то
![]()
где е0 – общая концентрация фермента в системе, известная из исходных данных и равная количеству фермента, загруженного в реактор. Теперь путем исключения (es) и е из трех последних уравнений можно получить уравнение (3.3), в котором Vmax=k2e0. Обычно, говорят, что реакция характеризуется уравнением Михаэлиса – Ментен, если ее скорость может быть описана уравнением (3.3), хотя в разработку и подтверждение правильности этого выражения внесли не меньший вклад и другие исследователи. Параметр vmax называют максимальной или предельной скоростью, a Km известна как константа Михаэлиса. Уравнение Михаэлиса – Ментен удачно описывает кинетику многих катализируемых ферментами реакций, но тем не менее оно не универсально. В последующих разделах и в упражнениях мы рассмотрим ряд модифицированных вариантов этого уравнения, которые точнее описывают конкретные ферментативные реакции, протекающие в специфических условиях.
Бриггс и Холдейн пришли к уравнению (3.3) другим путем, который, как было показано позднее путем экспериментального изучения кинетики и математического анализа, носит наиболее общий характер. Если реакция осуществляется в закрытом сосуде при энергичном перемешивании, то уравнения материального баланса для субстрата и фермент-субстратного комплекса можно записать следующим образом:
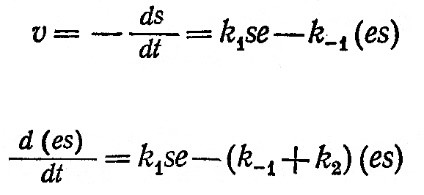
С учетом выражения (3.7) эти два уравнения представляют собой систему двух обычных дифференциальных уравнений с двумя неизвестными s и (es). Понятно, что соответствующие начальные условия при t=0 выражаются следующим образом:
![]()
Эти уравнения нельзя решить аналитически, но путем интегрирования с помощью компьютеров можно найти зависимости концентраций S, Е, ES и Р от времени. Полученные таким путем результаты приведены на рис. 3.8; очевидно, что после короткого стартового периода можно с весьма большой точностью принять, что
![]()
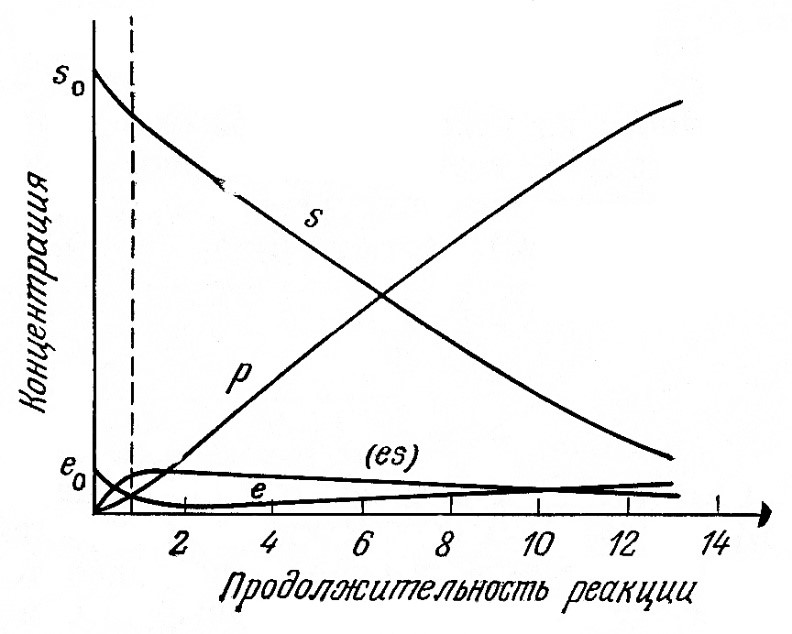
РИС. 3.8. Изменение концентраций веществ в реакции
Ключевым пунктом анализа Бриггса и Холдейна явилось допущение о правильности условия (3.8). Можно показать, что для нашего случая катализируемой ферментом реакции в закрытой системе это допущение, называемое обычно приближением квазиравновесного состояния, справедливо при условии достаточно малой величины отношения eo/so. Если же начальная концентрация субстрата сравнима с общей концентрацией фермента, то допущение (3.8) может оказаться неверным. В большинстве случаев, однако, количество ферментного катализатора намного меньше количества реагентов; в такой ситуации после стартового периода реакции выражение (3.8) можно считать применимым с достаточно высокой точностью.
Если далее, следуя рассуждениям Бриггса и Холдейна, из приведенных выше уравнений с помощью выражения (3.7) исключить е и (es), то мы придем к уравнению
![]()
Таким образом мы получили выражение типа уравнения Миха- злиса – Ментен, в котором
![]()
и
![]()
Обратите внимание на то, что здесь Km по физическому смыслу уже не является константой диссоциации.
Располагая математическим выражением для скорости реакции типа уравнения Михаэлиса – Ментен, мы можем определить изменения концентраций реагентов во времени путем интегрирования
![]()
Таким путем получаем, что
![]()
Разумеется, на практике это уравнение проще использовать для расчета времени t, необходимого для достижения определенных концентраций субстрата, а не наоборот.
Полезно сравнить ход реакции, предсказываемый уравнением (3.13), с результатами, полученными без приближения квазиравновесного состояния. Как показано на рис. 3.9, отклонения могут быть значительными, если общая концентрация фермента приближается к S0, поэтому в таких случаях не следует применять уравнение Михаэлиса – Ментен. На рис. 3.10 приведен другой пример, а именно зависимость эстеразной активности от концентрации фермента. Здесь предсказываемая моделью Михаэлиса – Ментен линейная зависимость справедлива при малых концентрациях фермента, но также не соблюдается при более высоких концентрациях. Запомните, что для линейного участка кривой тангенс угла наклона равен k2Sl(Km + S).
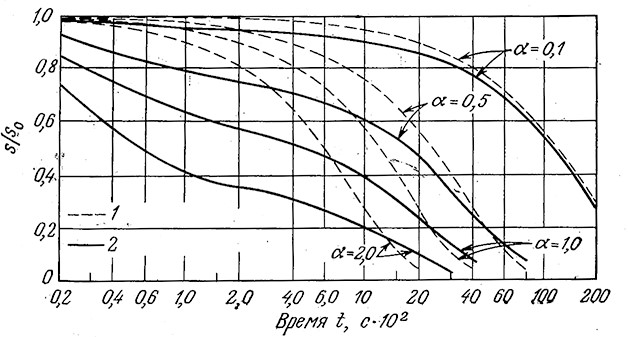
РИС. 3.9. Вычисленные зависимости степени гидролиза эфира ацетил-L-фенйл- аланина
химотрипсином от продолжительности реакции в реакторе периодического действия.
При больших величинах e0/s0= α наблюдается значительное несоответствие между точным решением
и решением в квазиравновесном приближении.
1 – решение в квазиравновесном приближении; 2 – точное решение,
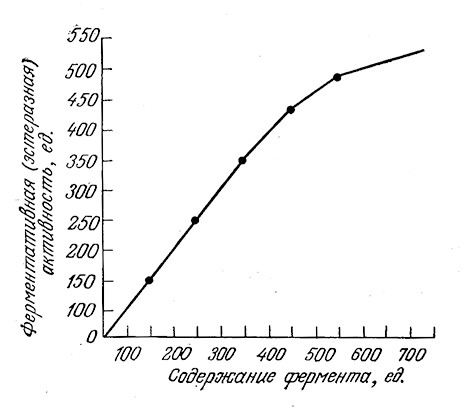
РИС. 3.10. При высоких начальных концентрациях фермента зависимость
эстеразной активности от концентрации эстеразы не подчиняется уравнению Михаэлиса – Ментен.
В некоторых случаях в зависимости от скоростей отдельных стадий реакции приближение квазиравновесного состояния может оказаться справедливым и при больших значениях e0/s0. В частности, такая ситуация могла бы возникнуть, если бы комплекс ES диссоциировал гораздо быстрее, чем образовывался, т. е. если бы Km была значительно больше S0. Однако обычно константа Михаэлиса очень мала и составляет от 10-2 до 10-5, поэтому такие случаи не типичны.
Отсюда следует, что если концентрация фермента сравнима с so, то, как правило, у нас мет достаточных оснований для упрощения модели кинетики соответствующего процесса с использованием приближения квазистационариого состояния. Относительная концентрация фермента может достигать больших значений, например, если работа ферментативного реактора продолжается и тогда, когда большая часть субстрата уже подверглась превращению. В таком случае s уменьшается до значений, сравнимых с е0, и на последнем этапе процесса уравнение Михаэлиса – Ментен может уже не отражать его реальной кинетики. К счастью, в этот период реакция резко замедляется и поэтому, как правило, уже не представляет практического интереса. Концентрация субстрата вблизи фермента может быть незначительной и при проведении ферментативных реакций на поверхности раздела фаз; реакции такого типа мы рассмотрим в разд. 4.4. В таких случаях следует учитывать и скорость переноса субстрата к границе раздела фаз. Ограничения, свойственные модели Михаэлиса – Ментен, выявились лишь в самое последнее время, и поэтому соответствующее уравнение все еще широко используется при изучении и проектировании ферментативных реакторов. Хотя в некоторых ситуациях равновесное приближение и уравнение Михаэлиса – Ментен нельзя считать достаточно точными, в целом они оказались очень полезными инструментами исследования, и в этом качестве мы часто будем их применять и в дальнейшем изложении.
Говоря о простом уравнении Михаэлиса – Ментен, нельзя не упомянуть, что точно таким же математическим выражением широко пользуются для оценки скоростей многих реакций, ускоряемых твердыми катализаторами. Уравнение (3.3) в химической технологии называется уравнением Ленгмюра – Хин- шелвуда или Хоугена – Уотсона. Поскольку в химической и нефтяной промышленности широко применяются реакции на синтетических твердых катализаторах, анализу и проектированию таких каталитических реакторов было посвящено огромное число работ. В большей части этих работ использовали уравнение Ленгмюра – Хиншелвуда; следовательно, их результаты с успехом могут быть перенесены и на катализируемые ферментами реакции, описываемые уравнением Михаэлиса– Ментен. В последующих главах мы еще не раз встретимся с аналогиями и сходством между классической химической и биохимической технологиями, но в то же время узнаем и многие особенности, характерные только для биологических процессов.
3.2.2. Определение параметров в уравнении Михаэлиса – Ментен
В своей первоначальной форме уравнение Михаэлиса – Ментен (3.3) неудобно для определения кинетических параметров Vmax и Km. Как показано на рис. 3.7, по графику зависимости V от s довольно сложно определить точное значение Vmax. Путем ряда несложных преобразований уравнения (3.3) нетрудно, однако, найти приведенные ниже уравнения, более пригодные для изображения результатов в графической форме и для графического определения параметров:
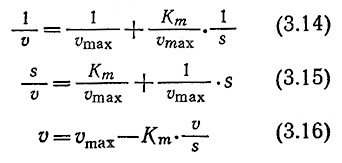
Каждое из приведенных уравнений отражает линейную зависимость одной величины от другой. В то же время при определении кинетических параметров по таким зависимостям следует принимать во внимание ряд свойственных им ограничений и недостатков. В графическом выражении уравнения (3.14) в виде зависимости 1/V от 1/s (известном под названием графика Лайнуивера – Бэрка) четко разделены зависимая и независимая переменные (рис. 3.11.а). Определяемые с наибольшей точностью значения скоростей реакций, близкие fmax, концентрируются возле исходной точки, а менее точно определяемые значения v удалены от нее и таким образом в наибольшей степени влияют на наклон прямой Km/ Vmax; поэтому в таких случаях нельзя применять метод наименьших квадратов. Второе уравнение (3.15) обеспечивает более равномерное распределение больших значений v и, таким образом, более точное определение наклона кривой 1/ Vmax, однако отрезок, отсекаемый прямой на координатной оси, часто слишком мал, что не позволяет достаточно точно определить этим методом Km- В третьем методе применяется так называемый график Эди – Хофсти, представляющий собой график зависимости v от v/s (рис. 3.11,6); здесь наименее точно определяемая переменная v является составной частью как функции, так и аргумента.
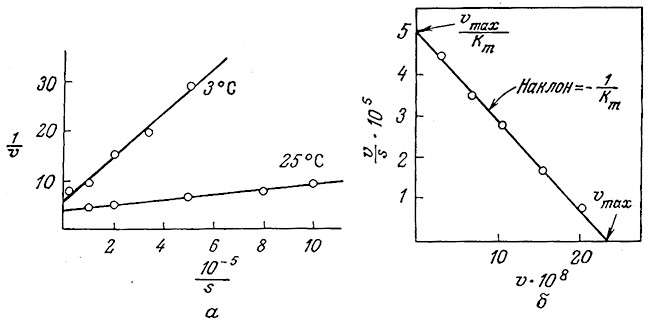
РИС. 3.11. а – результаты экспериментального изучения ферментативной активности пепсина в координатах Лайнуивера – Бэрка;
б – экспериментальные данные по изучению катализируемого химотрипсином гидролиза
метилового эфира дигидрокоричной кислоты в координатах Эди – Хофсти
Приведенные выше соображения диктуют следующую стратегию определения Vmax и Km. сначала определяют Vmax по графику – уравнения (3.14) (находят точную величину отрезка, отсекаемого прямой на оси 1/V) или уравнения (3.15) (точно определяют наклон прямой). Затем возвращаются к графику зависимости V от S и находят S1/2, т. е. такую концентрацию субстрата, при которой V равно Vmax/2. Как мы уже упоминали при обсуждении уравнения (3.3), Km по величине и размерности равно S1/2.
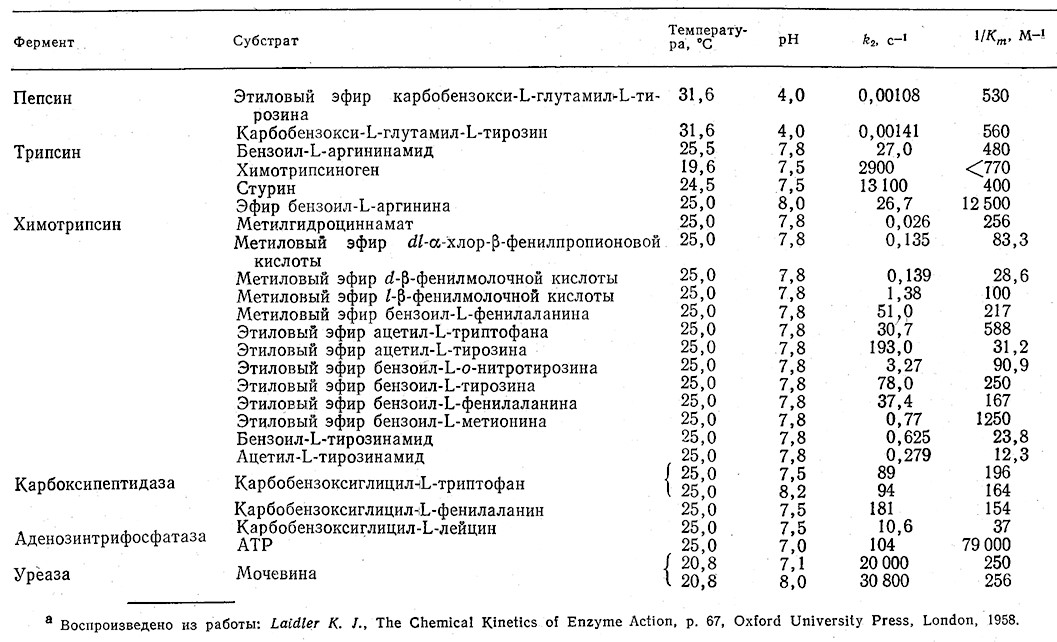
Важно представлять себе, в каких диапазонах могут изменяться эти кинетические параметры. В табл. 3.4 приведен ряд параметров, характерных для различных ферментов. Обратите внимание на то, что величина k2, может изменяться в очень широких пределах, a Km, напротив, обычно имеет значения 2•10-3–10•10-3 М. Почти все приведенные в табл. 3.4 данные были получены при умеренной температуре и почти нейтральном рН. Исключение составляет пепсин, основная биологическая функция которого сводится к гидролизу белков в кислой среде желудка и который поэтому наиболее активен при низких значениях рН; эти значения, естественно, использовались и при экспериментальном определении его кинетических параметров. Модели, отражающие влияние рН й температуры на кинетику ферментативных реакций, мы рассмотрим в разд. 3.6.
3.2.3. Кинетика обратимых реакций, реакций с двумя субстратами
и с активацией фермента кофактором
Равновесие многих катализируемых ферментами реакций, например, реакций гидролиза биополимеров, сильно смещено в Сторону продуктов реакции, поэтому, как правило, такие превращения можно считать необратимыми. В других случаях, например, при изомеризации глюкозы во фруктозу под действием фермента глюкозоизомеразы, может установиться равновесное состояние, требующее учета вклада обратной реакции. В качестве простейшей модели обратимой ферментативной реакции рассмотрим кинетику последовательных превращений типа
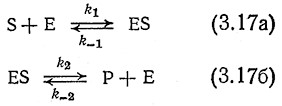
Эта последовательность элементарных реакций отличается от последовательности реакций, предложенных Анри – Михаэ- лисом – Ментен и выражаемых уравнениями (3.4), только тем, что уравнение (3.17Б) теперь предусматривает образование комплекса ES из продукта реакции и свободного фермента.
Таблица 3.4. Кинетические параметры некоторых ферментативных реакций
Как и ранее, будем считать, что реакционная смесь находится в закрытом сосуде и эффективно перемешивается. Тогда на основе материального баланса различных форм фермента [уравнение (3.7)] с учетом приближения квазистационарного состояния [уравнение; (3.8)] легко находим, что
![]()
Здесь Vs и Ks аналогичны Vmax и Km. в уравнениях (3.10) и (3.11) соответственно, и
![]()
Подавляющее большинство катализируемых ферментами реакций протекает с участием по меньшей мере двух субстратов. В то же время, чаще всего| одним из субстратов является вода, концентрация которой практически постоянна и обычно в 1000 или более раз превышает концентрации других субстратов. Как мы покажем позднее в этом же разделе, в таких случаях можно считать, что реакция протекает с одним субстратом S, и, следовательно, ее кинетику можно изучать так, как это было описано в предыдущем разделе. Кроме того,, описанные ниже кинетические модели иногда могут объяснять и влияние кофакторов на скорости ферментативных реакций.
По всей вероятности, во многих процессах с участием двух субстратов могут образовываться тройные комплексы, в которых фермент одновременно связан с двумя субстратами. В таком случае возможна, например, такая последовательность реакций:

Как и раньше, строчными буквами мы будем обозначать концентрации, а такими же буквами в скобках – концентрацию одной формы, например, фермент-субстратного комплекса. Если допустить, что первые четыре реакции (3.20) равновесны, то получим
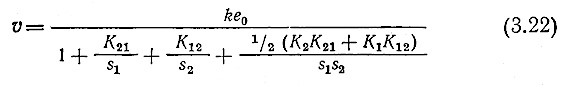
Вывод этого выражения не представляет затруднений, если учесть, что общая концентрация фермента е0 должна быть равна сумме концентраций свободного фермента е и трех комплексов ES1, ES2 и ES1S2. Как и в случае реакций с одним субстратом, в ферментативных реакциях с двумя субстратами можно применить приближение квазиравновесного состояния. В общем случае, однако, в результате получается довольно громоздкое уравнение с таким числом параметров, что оно становится непригодным для практического использования. Удовлетворительное по ряду параметров приближение описывается уравнением (3.22).
Уравнение (3.22) можно далее несколько упростить, если принять во внимание, что условия равновесия требуют, чтобы
![]()
Соответствующим преобразованием уравнения (3.22) прийти к уже знакомой форме уравнений:
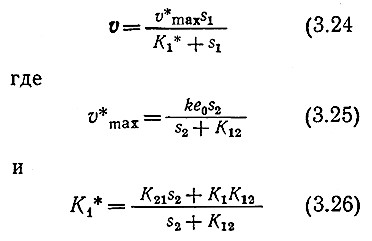
Из последних трех уравнений следует, что если S2 постоянна, а S1 изменяется, то реакция будет подчиняться уравнению Михаэлиса – Ментен. В то же время уравнения (3.25) и (3.26) показывают, что кажущиеся максимальная скорость и константа Михаэлиса зависят от концентрации S2.
Если считать приведенные выше последовательность реакций и выражение для скорости реакции с двумя субстратами правильными, то мы можем проверить справедливость уравнения Михаэлиса – Ментен (3.3), допустив, что один субстрат находится в большом избытке. Тогда V *max становится равной ke0, a K1* приближается к постоянному значению K21. Следовательно, реакцию с двумя субстратами при S2 >> K12 можно рассматривать как реакцию с одним субстратом.
На рис. 3.12 изображена схема механизма ферментативной реакции с одним субстратом (или с двумя субстратами при S2 >> K12) при участии кофактора, которым может быть ион металла или кофермент. Поскольку эта ситуация аналогична только что рассмотренному нами двухсубстратному механизму, здесь нет необходимости снова обращаться к допущению о равновесных последовательных реакциях. Если принять, что субстрат связывается только апофермент-коферментным комплексом, то в конце концов мы получим
где с – концентрация кофактора. Если считать концентрацию субстрата s постоянной, то это выражение преобразуется в уравнение Михаэлиса – Ментен, отражающее зависимость скорости реакции от концентрации кофактора с. Так, при невысокой концентрации кофактора (c << Kc) реакция будет иметь первый порядок по концентрации кофактора с. С другой стороны, при С >> Кс мы получим уравнение скорости реакции с одним субстратом, которая не зависит от концентрации кофактора.
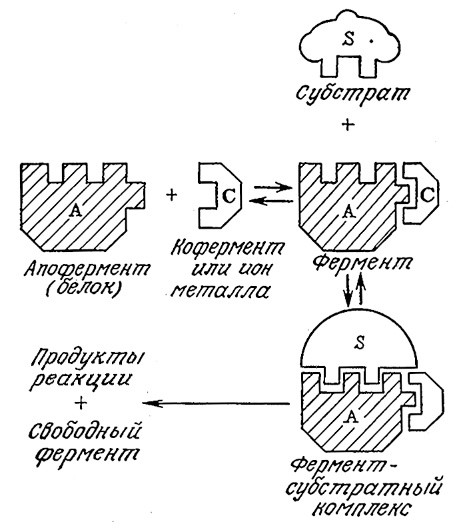
РИС. 3.12. Схематическое изображение предполагаемого механизма
ферментативного катализа с участием кофактора.
Если S1 и S2 или С и S1 связываются с ферментом в строго определенном порядке, то соответствующее уравнение скорости процесса можно получить, приняв, что Кij запрещенной реакции приближается к бесконечно большой величине.
