В разд. 3.2.2 мы рассмотрели различные графические способы определения параметров Vmax и Km, входящих в уравнение Михаэлиса – Ментен. Как показывает анализ Бриггса – Холдейна, эти параметры определяются константами скоростей элементарных стадий реакции k1, k-1 и k2. В то же время из уравнений (3.10) и (3.11) следует, что знания параметров Km и vmax еще недостаточно для определения скорости отдельных стадий реакции. Для оценки последних, очевидно, необходимо найти какие-то иные, независимые взаимосвязи между константами скоростей элементарных стадий и экспериментальными данными.
Такие более фундаментальные кинетические параметры представляют интерес по меньшей мере по двум причинам. Во-первых, они дают углубленное представление о реальных процессах, происходящих в ходе катализируемых ферментами реакций. Мы можем узнать, например, насколько быстро субстрат взаимодействует с ферментом, и сравнить эти величины со скоростью обратного процесса диссоциации комплекса ES. Во-вторых, как мы уже видели, упрощения, лежащие в основе уравнения Михаэлиса – Ментен, с достаточной точностью оправдываются только при относительно невысоких концентрациях фермента. Если это условие не соблюдается, то изучение кинетики ферментативной реакции возможно только при учете данных материального баланса для s, р и (es), для чего в свою очередь необходимо знать константы отдельных стадий реакции k1, k-1 и k2. По указанным причинам нам представляется целесообразным рассмотреть здесь экспериментальные методы определения этих параметров и полученные с помощью таких методов результаты.
В методе кинетики предстщионарного состояния основное внимание уделяется короткому периоду (около 1 с или меньше) сразу после начала реакции, когда приближение квазистационарного состояния еще неприменимо. В течение этого периода концентрация субстрата изменяется незначительно, что позволяет приближенно решить уравнения материального баланса для (es) и р. Сравнение найденной таким образом зависимости р от t с экспериментальными данными, а также найденные значения Vmax и Km. дают всю информацию, необходимую для расчета k1, k-1 и k2. Экспериментальной основой этого метода является методика остановленного потока, разработанная и развитая Б. Чансом с сотрудниками. Согласно этой методике, растворы фермента и субстрата быстро смешивают в кювете спектрофотометра; спектрофотометрический метод позволяет контролировать изменения концентраций исходных веществ и продуктов реакции, осуществляющиеся в течение нескольких миллисекунд.
Разработанные и широко применявшиеся Эйгеном и сотрудниками релаксационные методы дают возможность изучать реакции, завершающиеся в течение нескольких наносекунд. Существует несколько вариантов этого метода, но все они основаны на принципе возмущения равновесного (или стационарного) состояния реакционной смеси путем искусственного скачкообразного изменения некоторых условий реакции, например, температуры, давления или напряженности электрического поля. Последующий отклик реакционной системы на это изменение регистрируют непрерывно. Как схематически показано на рис. 3.13, отклик на скачкообразное изменение условий реакции представляет собой не что иное, как переход в новое, почти равновесное или стационарное состояние.

РИС. 3.13. В релаксационных методах небольшое скачкообразное изменение условий реакции,
например: температуры, вызывает быстрый переход в новое равновесное состояние.
3.3.1. Релаксационные методы изучения кинетики
В этом разделе мы несколько подробнее остановимся на релаксационных методах, основанных на скачкообразном изменении условий реакции. Теория и практика релаксационных методов была разработана также в применении к осциллирующим возмущениям условий реакций. Сначала рассмотрим равновесие только между субстратом, ферментом и фермент-субстратным комплексом:
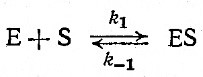
Мы знаем, что для этой реакции
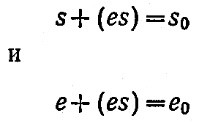
Поэтому для эффективно перемешиваемой реакционной смеси, находящейся в реакторе периодического действия, уравнение материального баланса для s
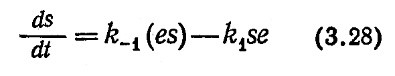
принимает форму
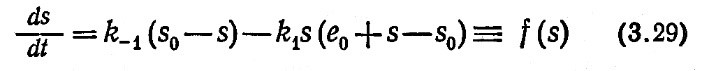
Если мы символом s* обозначим концентрацию s в равновесном состоянии после скачкообразного изменения условий реакции, то s* можно определить, решив уравнение
![]()
где f(s) – правая часть уравнения (3.29), равная ds/dt. В любом релаксационном эксперименте s будет близко s* и приближенно можно считать, что f(s) равно первым членам в разложении Тейлора типа
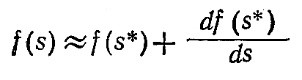 + члены порядка (s–s*)2 и высших пррядков. (3.31)
+ члены порядка (s–s*)2 и высших пррядков. (3.31)
Здесь f(s), согласно уравнению (3.30), равно нулю. Обозначив символом χ отклонение от равновесной концентрации
![]()
и принимая во внимание, что s* не зависит от времени, путем преобразования уравнений (3.29)–(3.32) можно получить линейное уравнение следующего вида [если пренебречь членами второго и высшего порядков в уравнении (3.31)]:
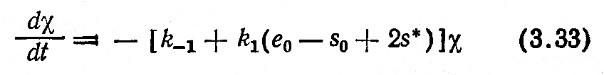
Если система сначала находится в состоянии исходного равновесия, которое отвечает условиям, предшествующим скачкообразному возмущению, то
![]()
а затем
![]()
где
![]()
В уравнении (3.36) е* выражает концентрацию не связанного в комплекс фермента в состоянии конечного равновесия. Уравнение (3.35) свидетельствует о том, что τ можно определить по наклону прямой, отражающей полулогарифмическую зависимость χ(t) от ∆χ0. Другими словами, τ представляет собой время, в течение которого χ(t) уменьшается до 37% от своего первоначального значения. Определив τ, χ* и е* (или e0 и S0), далее по уравнению (3.36) можно найти взаимосвязь между k1 и k-1 не зависящую от уравнения равновесия. Таким путем можно вычислить как k1 так и k-1.
Этот метод нашел широкое применение в химической кинетике, в том числе и в кинетике рассматриваемого здесь ферментативного катализаи
3.3.2. Некоторые результаты изучения кинетики переходных состояний
В табл. 3.5 приведены значения k1 и k-1 для ряда ферментов и субстратов. Эти данные были получены с помощью описанных выше методов, а также ряда других специальных методик, описанных в приведенной в конце настоящей главы литературе. Нетрудно видеть, что константы скорости прямой реакции субстрата с ферментом k1 почти всегда близки к теоретическому максимальному значению, составляющему для абсолютной скорости бимолекулярных соударений в растворе от 109 до 1011 [(моль•с)/л]-1. Напротив, обратный процесс протекает сравнительно медленно, и его скорость, как и скорость диссоциации промежуточного фермент-субстратного комплекса на продукт реакции и свободный фермент (см. значения k2 в табл. 3.4), в значительно большей степени зависит от природы реагирующих веществ.
Таблица 3.5. Константы скорости некоторых элементарных стадий фермент-субстратных реакцийa

Описанными выше методами была выявлена другая интересная особенность ферментативного катализа, заключающаяся в образовании нескольких промежуточных фермент-субстратных комплексов ES. В ряде случаев комплекс претерпевает ряд последовательных конформационных превращений, которые можно рассматривать как реакции изомеризации. Методами изучения стационарных состояний невозможно установить, образуется ли одно или несколько промежуточных соединений, если скорость диссоциации последнего промежуточного комплекса (до конечных продуктов реакции) мала по сравнению со скоростью превращения комплексов, образующихся на предыдущих стадиях. В таких случаях ценная информация может быть получена релаксационными методами.
