ОБЩИЕ СВЕДЕНИЯ
Несмотря на длительное и интенсивное изучение, многае аспекты патогенеза сальмонеллеза остаются невыясненными. В изучении этого вопроса можно выделить несколько этапов:
1) установление этиологической роли сальмонелл в развитии пищевых отравлений;
2) доказательство значения эндотоксина сальмонелл в патогенезе заболевания;
3) изучение роли вирулентных свойств сальмонелл в развитии заболевания;
4) исследование молекулярных механизмов воздействия токсинов возбудителя на микроорганизм.
Течение сальмонеллеза характеризуется широким диапазоном клинических проявлений – от бессимптомного до тяжелой септицемии. Наиболее частой является гастроинтестинальная форма сальмонеллеза. Причина столь широкого полиморфизма клинических вариантов заболевания не ясна по сей день. Можно лишь предположить, что это связано с вирулентностью возбудителя и состоянием иммунореактивности макроорганизма. Как уже отмечалось, основным фактором патогенное сальмонелл является эндотоксин. Однако для того, чтобы эндотоксин проявил свое патогенное действие, должно пройти несколько этапов развития инфекционного процесса:
а) колонизация (заселение) возбудителем участка тела в месте внедрения;
б) инвазия во внутреннюю фазу с последующим размножением;
в) гибель возбудителя и высвобождение эндотоксина.
По неизвестным причинам инфекционный процесс может остановиться на этапе колонизации, а инвазия может ограничиться близлежащими тканями. Вероятно, это имеет место в большинстве случаев, что и приводит к развитию гастроинтестинальной формы сальмонеллеза [Войно-Ясенецкий М. В., 1981; Ющук Н. Д., 1980; Блюгер А. Ф. и др., 1975; Giannella, 1979].
Признание инициирующей роли сальмонелл в развитии заболевания в свою очередь поставило вопрос о механизмах, обеспечивающих проникновение сальмонелл через барьер энтероцитов. Известно, что на поверхности, обращенной в просвет кишечника, энтероциты имеют специальные образования – микроворсинки, от 1000 до 3000 на каждую клетку. Микроворсинки соединены между собой гликопротеином, аналогичным гликокаликсу, покрывающему свободные поверхности других клеток, микробных в том числе. Гликокаликс содержит ряд ферментов, обеспечивающих всасывание. Существует мнение, что гликокаликс выполняет и защитную функцию, препятствуя проникновению эндотоксина и микробов во внутреннюю среду организма [Войно-Ясенецкий М. В., 1981]. Кроме того, энтероциты в апикальной части скреплены замыкательным комплексом, обеспечивающим тесную связь между соседними клетками.
Эти сведения важны для понимания структурных основ защиты внутренней среды организма от многочисленных микроорганизмов, попадающих с пищей. Поэтому для развития первой фазы патогенеза сальмонеллеза важное значение имеют факторы, нарушающие структурно-функциональное состояние желудочно-кишечного тракта, хронические заболевания желудочно-кишечного тракта, дисбактериоз, гиповитаминоз и др. [Блюгер А. Ф. и др., 1975]. Указанные состояния, видимо, способствуют развитию сальмонеллеза и при небольшом количестве бактерий в продуктах питания. По мнению М. В. Войно-Ясенецкого (1981), лишено основания предположение о том, что сальмонеллы проникают через щеточную каемку в захвативших их гранулоцитах или макрофагах. Поэтому существует предположение о наличии у сальмонелл особых свойств, позволяющих им проникать сквозь барьер энтероцитов.
ВНЕДРЕНИЕ ВОЗБУДИТЕЛЯ
Из исследований на добровольцах известно, что развитие сальмонеллеза определяется дозой живого возбудителя [McGullough N. В., Eisele С. W., 1951]. Эти наблюдения согласуются с положениями инфектологии, касающимися инфекционного процесса, вызванного условно-патогенной флорой, и свидетельствуют о значении пищевого фактора в передаче инфекта при сальмонеллезе, так как он обеспечивает поступление максимального числа бактерий в организм [Блюгер А. Ф. и др., 1975].
Проникновение сальмонелл в слизистую оболочку тонкой кишки осуществляется в два этапа. Вначале бактерии преодолевают эпителиальный барьер. По данным Takeuchi A., Sprinz Н. (1967), под действием возбудителя сначала повреждаются микроворсинки и десмосомы, соединяющие отдельные эпителиальные клетки между собой. Затем бактериальные клетки, повреждая апикальную цитолемму, образуют в ней инвагинацию. Эта инвагинация превращается в вакуоль, которая окружается плотным материалом, в состав которого входят митохондрии, цистерны эндоплазматической сети и рибосомы. Продолжительность первой стадии фазы внедрения возбудителя в эксперименте достигает 24 ч от повреждения щеточной каемки (через 12 ч после заражения) до полного преодоления эпителиального барьера.
Во вторую стадию бактерии проникают в собственную пластинку слизистой оболочки [Takeuchi A., Sprinz Н., 1967]. Здесь их захватывают макрофаги. Полагают, что в этот период сальмонеллы способны размножаться и выделять хемотаксический фактор. Видимо, благодаря выделению последнего в эту стадию в слизистой оболочке появляется большое количество полиморфно-ядерных лейкоцитов. Бактериемия в эксперименте выявлена через 72 ч после заражения животных.
Принято считать, что патогенность сальмонелл, т. е. их способность к внедрению, определяется прежде всего способностью к адгезии и колонизации [Полоцкий Ю. Е., Авдеева Т. А., 1981].
Достижения биологии и возникновение новой дисциплины – мембранологии – способствовали развитию новых представлений о явлениях, характеризующих взаимодействие микроба-возбудителя с клетками микроорганизма. На первом этапе инфекционного процесса при сальмонеллезе взаимодействие двух клеток – микробной (прокариот) и энтероцита (эукариот) – начинается с биололического узнавания. Специфическое узнавание друг друга молекулами и клетками является, по современным представлениям, основополагающим принципом функционирования всех биологических систем. Не является исключением и инфекционный процесс, в том числе прикрепление (адгезия) бактерий к клеткам-мишеням, последующее заселение (колонизация) тканей, фагоцитоз, развитие специфической иммунореактивности.
В свете последних данных можно предполагать, что dtoii внедрения сальмонелл в энтероциты обеспечивается системой биологического узнавания – лнганд-рецептор [Кзенчук Ю. В., 1985; Maas W. К., 1981]. Под лигандами ноиимают особые структуры бактериальной клетки, обеспечивающие специфическое взаимодействие с клетками макроорганизма. Лиганд – рецепторное узнавание – предполагает существование на поверхности клетки особых рецепторов, способных принять поступающий сигнал, трансформировать его в определенное действие клетки, объединить сигнал р действие клетки.
Распознающая часть рецептора обычно располагается па внешней стороне мембраны клетки, а эффекторная может располагаться вне мембраны, в мембране или в ее части, обращенной к цитоплазме. Помимо лиганд-рецепторного взаимодействия микробной клетки и энтероцита, этот феномен могут дополнять ван-дер-ваальсовы силы, силы электростатического взаимодействия, водородные связи и гидрофобные силы [Езепчук Ю. В., 1985]. При атом лиганд-рецепторный контакт совместно с гидрофобными взаимодействиями направлен на преодоление электростатического отталкивания между микробом и энтероцитом.
В бактериальной клетке функцию распознавания и прикрепления обеспечивают особые компоненты стенки – адгезины.
Адгезины взаимодействуют с поверхностью эукариотической клетки по типу группы веществ растительного происхождения, названной лектинами. Лектины способны избирательно осаждать группоспецифические вещества крови и агглютинировать эритроциты. В основе этого феномена и действия адгезинов бактерий лежит один и тот же принцип углевод-белкового распознавания, что и послужило основанием для определения адгезинов как бактериальных лектинов [Езепчук Ю. В., 1985; Stendahl О., 1982]. Долгое время считалось, что адгезины строением напоминают фимбрии (пили). Действительно, в значительном количестве работ установлено участие этих образований в процессе прикрепления бактерий к клеткам тканей человека и животных. Вместе с тем у сальмонелл найдены пили, но участвующие в процессе адгезии (пили II, III типов). У сальмонелл также обнаружены особые фибриллы, выполняющие функции адгезинов. У сальмонелл фибриллярные адгезины встречаются наряду с фимбриями [Burrows М.–R. et al., 1976]. Следует отметить, что в процессе адгезии сальмонелл важную роль играет их липополисахаридный комплекс (ЛПС) [Shands J. W., 1973; Beachey E. H. et al., 1979]. Таким образом, лиганд-рецепторное взаимодействие является начальным этапом внедрения сальмонелл во внутреннюю среду организма, оно же определяет специфичность выбора поражаемой ткани [Езепчук Ю. В., 1985; Jones G. W., 1980].
Детали строения рецепторов макроорганизма, обеспечивающих взаимодействие с микроорганизмами, к настоящему моменту мало изучены, но их значение для развития инфекционного процесса исключительно велико [Deneсе К., 1983]. Именно лектиеы клеток макроорганизма благодаря специфичности строения и конфигурации их углеводных компонентов обеспечивают определенный тропизм возбудителя [Hubbe М. А., 1981; Glancy J. et al., 1981; Aning К. F. et al., 1983].
Детальная классификация лектинов микроорганизмов пока не создана. Установлено, что патогенные бактерии взаимодействуют с клетками животных не по отдельности, а большим числом. По этой причине наблюдается конкурентное прилипание–бактериальная интерференция [Botta G. А., 1981; Montakhab М. et al., 1981; Willshaw G. A. et al., 1982; Broughton R. A. et al., 1983]. У многих бактерий адгезия угнетается на 60–98% Д-^маннозой. С помощью электронной микроскопии установлено, что активность лектинов, подавляемая Днманнозой, локализована на белковых субъединицах жгутиков (молекулярная масса 38 500 и З6 000 D). Лектины гомологичны [Suegara N. et al., 1981]. Некоторые из этих белков устойчивы к перевариванию трипсином, что имеет определенное значение в заражении кишечными инфекциями [Inman L. R. et al., 1983].
Размножение сальмонелл, приводящее к увеличению их популяции и к заселению поражаемой ткани, определяется двумя факторами: адгезинами микроба и состоянием субстанции, защищающей бактериальную клетку от фагоцитоза. Способность бактерий, в том числе и сальмонелл, образовывать поверхностные структуры (пили) коррелирует с их вирулентностью [Овод В. В., Вершигора А. Е., 1982; Костюкова Н. Н., Миронова Т. К., 1983]. Вместе с тем потеря бактериальной клеткой этих свойств ведет к утрате одного из факторов вирулентности, связанного с адгезивностью бактерий [Полоцкий Ю. А., Авдеева Т. А., 1980; Ткаченко В. В., 1982; Реагсе A. W., Buchanan М. Т., 1980].
Наши исследования показали, что повышенные концентрации высших жирных кислот в питательной среде вызывает торможение роста сальмонелл. Видимо, натриевые соли высших жирных кислот, как мыла, обладают свойствами поверхностно-активных веществ. Нарушая проницаемость мембран бактериальной клетки, они вызывают замедление ее роста [Работнова И. Л. и др., 1981; Перт Дж., 1978].
Известно, что сальмонеллы не подвергаются фагоцитозу в макрофагах [Зуфаров К. А. и др., 1974].
В макрофагах сальмонеллы не только сохраняются, но и могут размножаться [Войно-Ясенецкий М. В., 1981]. И макрофагах сальмонеллы попадают в лимфатические узлы и даже в кровь. Однако предполагают, что в большинство случаев сальмонеллы проникают в кровь через слизистую оболочку тонкой кишки.
По данным Ю. Я, Тендентника и соавт. (1980), сальмонеллам присущи высокие инвазионные свойства, благодаря которым они способны проникать в кровь уже через несколько минут после Церорального или же внутрикишечного заражения. Причем считают, что при нероральном заражении основная масса сальмонелл сохраняется в глотке и легких, а при кишечном – в месте их введения, в мезенториальных узлах и печени. По мнению авторов, именно из печени (основном месте накопления сальмонелл) и меле птериальных лимфатических узлов и происходит вторичная диссеминация возбудителя. Установлено, что сальмонеллы, остающиеся в просвете кишки, погибают. При этом их количество уменьшается в тысячу раз уже к исходу 30-й минуты опыта.
Принято считать, что в собственном слое слизистой оболочки тонкой кишки идет интенсивное разрушение бактерий, в процессе которого происходит высвобождение эндотоксина. Всасывание последнего в кровь, а также его действие на энтероциты знаменуют собой начало клинических проявлений сальмонеллеза. В процессе бактериемии и последующей фиксации микробов в клетках макрофага л ыю-гистиоцитарной системы сальмонеллы разрушаются и при этом высвобождаются новые порции эндотоксина.
Бактериемия у больных сальмонеллезом встречается часто, по обычно бывает кратковременной. В эксперименте показино, что бактериемия носит перемежающийся характер. Это объясняется чередованием размножения сальмонелл в макрофагах и последующим их выходом в кровь [Поставит В. А., 1984].
Быстрая гибель сальмонелл в собственном слое слизистой оболочки кишечника, а также фиксация сальмонелл в макрофагально-гистиоцитарной системе с последующим их быстрым разрушением характерны для гастроинтестинальной формы сальмонеллеза. Вместе с тем в незначительном проценте случаев в местах фиксации сальмонелл могут формироваться очаги пролиферативного, реже гнойного, воспаления, что характерно для развития соответственно тифоидной и септической форм сальмонеллеза. Причины этого не совсем ясны. Предполагают, что развитие тифоидных и септических форм сальмонеллеза определяется возможным снижением чужеродности антигенов для иммунокомпетентных клеток или же развитием вторичного иммунодефицита [Блюгер А. Ф. и др., 1975]. Действительно, как уже указывалось, набор антигенов у сальмонелл не является стабильным, возможно развитие антигенных вариаций, что в ряде случаев и способствует появлению феномена антигенной мимикрии, в результате которой у сальмонелл могут появиться общие с макроорганизмом антигенные детерминанты, снижающие иммунный ответ организма.
Именно толерантность, приобретенная за счет антигенной мимикрии у сальмонелл, вероятно, лежит в основе острой или хронической форм субклинического течения сальмонеллезной инфекции [Блюгер А. Ф. и др., 1975].
МЕХАНИЗМЫ ВОЗДЕЙСТВИЯ ЭНДОТОКСИНА САЛЬМОНЕЛЛ НА МАКРООРГАНИЗМ
Эндотоксин и гомеостаз
В настоящее время накоплены обширные данные в отношении характеристики различных патологических изменений, возникающих под влиянием эндотоксина в организме человека или экспериментального животного.
Детальное изучение эффектов действия эндотоксина показало, что многие реакции развиваются после определенного скрытого периода, ряд из них не воспроизводится в условиях прямого контакта эндотоксина с клетками и тканями [Шенкман Б. 3., 1985].
При анализе возможных механизмов патогенного действия эндотоксина необходимо прежде всего иметь в виду огромное количество вызываемых им реакций со стороны всех органов и систем.
Анализ данных литературы убеждает в том, что патогенез сал ьмоисллезной интоксикации связывают прежде всего с представлениями об эндотоксине, который благодаря своим токсичным свойствам вызывает различные нарушения гомеостаза. Большинство современных исследовав пин характеризуется существенной недооценкой макроорганиама, роли его адаптационно-приспособительных механизмов в патогенезе заболевания.
По мнению большинства исследователей, гибель сальмонелл происходит в верхнем отделе желудочно-кишечного тракта, о чем свидетельствуют результаты иммуноморфологнческих исследований, выявивших накопление большого количества пммунокомпетентных клеток в собственном слое слизистой оболочки, в том числе макрофагов, что не характерно для других острых кишечных инфекций. Установлена также преимущественная выработка секреторных иммуноглобулинов энтероцитами начального отдела тонкой кишки. Гибель сальмонелл и высвобождение эндотоксина происходят преимущественно в мезентериальных лимфатических узлах и крови.
Поскольку именно расшифровка механизмов действия эндотоксина привела к развитию принципиально нового подхода к терапии сальмонеллезов, мы считаем необходимым более подробно рассмотреть механизмы срыва гомеостаза, возникающего под действием эндотоксинов сальмонелл. Действие эндотоксинового комплекса на организм определяется входящим в его состав липополисахаридом. По-видимому, липополисахарид взаимодействует с большинством клеток макроорганизма. Однако специфически реагируют на его воздействие лишь те клетки, которые обладают рецепторами к эндотоксину (лиганд-рецепторное распознавание). Взаимодействие липополисахарида с лини дами, входящими в состав клеточной мембраны, представляет собой сложную многокомпонентную химическую реакцию, сущность которой до настоящего времени остается неясной [Morrison D. С., Rudbach J. А., 1981].
Вместе с тем, по мнению некоторых исследователей, для молекулы эндотоксина грамотрицательных бактерий в организме имеются как внеклеточные, так и связанные с мембранами различных клеток'рецепторы. В частности, по данным G. F. Springer, J. С. Adye (1975), такие рецепторы имеют форменные элементы крови: тромбоциты, лейкоциты и лимфоциты. Из цитоплазматических мембран этих клеток выделены глицерофосфатиды, способные связываться с эндотоксином. Из эритроцитов выделен липогликопротеид, обладающий таким же свойством. Из глицерофосфатидов клеточных мембран наибольшим сродством к эндотоксину обладал фосфатидилхолин.
Результат лиганд-рецепторного взаимодействия эндотоксина оценивается неоднозначно. При изучении воздействия бесклеточных фильтров 50 различных серотипов сальмонелл на культуру клеток почек зеленых мартышек было установлено, что токсин вызывает отслоение 20-50% клеток и торможение синтеза белка [Коо F. G., Peterson J. W., 1983]. По мнению этих авторов, именно торможение синтеза белка является первичным эффектом действия токсина.
В то же время существует предположение, что липополисахарид проникает в клетки с помощью эндоцитоза и вызывает здесь цепь событий, приводящих к повреждению [Bradley S. G., 1979]. Видимо, липополисахарид, связываясь с внутренней мембраной митохондрий, активирует гликолиз. Кроме того, появляются пероксидные радикалы и пероксид водорода, оказывающие повреждающее действие на органеллы клетки и вызывающие выброс гидролаз из лизосом.
Известна убиквитарность воздействия сальмонеллезного эндотоксина на организм. Однако результаты этого воздействия на различные органы и системы неоднозначны. Так, наиболее выраженным, по-видимому, является действие эндотоксина на секрецию электролитов и жидкости в тонкой кишке, ее резкое усиление – один из основных клинических признаков сальмонеллеза [Pauwell D. N. et al., 1971]. Одновременно развивается диссеминированное внутрисосудистое свертывание крови – следствие воздействия эндотоксина на свертывающую систему крови fElin R. J., Wolf М. S., 1976].
В настоящее время мнения о механизме диареи расходятся. R. A. Giannella и соавт. (1973) связывают развитие диареи прежде всего с пенетрацией слизистой оболочки возбудителем. Большое значение R. A. Giannella (1979) придает также развитию под действием сальмонелл острого воспаления ® слизистой оболочке тонкой кишки. Полагают, что именно в связи с острым воспалением в тонкой кишке резко возрастает уровень секреторных процессов. При этом проницаемость слизистой оболочки не нарушается [Kinsey М. D. et al., 1976].
Вместе с тем имеются убедительные экспериментальные данные об участии эндотоксина сальмонелл в повышении проницаемости слизистой [Арсеньева Л. С., 1981].
При внутрибрюшинном введении эндотоксина сальмонелл кроликам было отмечено резкое уменьшение соотношения холестерин/фосфолипид в щеточной каемке энтероцитов слизистой оболочки. Учитывая тот факт, что проницаемость зависит от состава фосфолипидов и соотношения холестерин/фосфолипид, сделан вывод о значительном повышении проницаемости мембран щеточной каемки.
Механизм активации секреции электролитов и жидкостей при сальмонеллезе сложен и во многом неясен. Безусловно, важна в этом процессе роль различных физиологически активных веществ. Действительно, разнообразные медиаторы воспаления обеспечивают реакцию микроциркуляторного русла, а также вызывают повышение его проницаемости, усиливают хемотаксис. Среди тканевых (клеточных) медиаторов воспаления важное место занимают иростагландины, обладающие широким спектром фармакологического действия, направленность которого зависит от их химического строения. Среди плазменных медиаторов важны компоненты кининовой системы, а также систем свертывания крови и комплемента.
Часть медиаторов оказывает непосредственное (прямое) действие на клетки, а другие – опосредованное. Медиаторы прямого действия образуются при воспалении очень рано. К ним относятся гистамин, серотонин, некоторые простагландины. В первую фазу повышения проницаемости сосудистого русла, которая развивается сразу после повреждения, наибольшее значение имеют пистамин, серотонин. Через несколько часов развивается вторая фаза, которая очень разнообразна по механизмам развития. Главную роль в этой фазе играют компоненты калликреин-кининовой системы, простагландины, а также лейкокинны. Появление последних связано с деятельностью полиморфно-ядерных лейкоцитов.
В настоящее время выявлены признаки воздействия эндотоксина почти на все механизмы регуляции гомеостаза. Видимо, можно предполагать как повреждающее действие эндотоксина на клетки отдельных органов и систем (энтероциты, гепатоциты, тромбоциты), так и стимулирующее или тормозящее его действие на соответствующие рецепторы клеточных мембран. Последний механизм, видимо, более характерен для систем, осуществляющих регуляцию гомеостаза гуморальным путем – с помощью гормонов и физиологически активных веществ.
Регуляция функций на уровне систем, органов, тканей и отдельных клеток обеспечивается химическими факторами, которые представляют одну из форм нейрогуморальной регуляции.
Важнейшую группу среди химических факторов составляют физиологически активные вещества, к которым относят циклические нуклеотиды, ангиотензины, кинины, катехоламины, компоненты каллшсреинкининовой системы, простагландины. В регуляции уровня выделения физиологически активных веществ (простагландинов, кининов) велико значение циклического аденозинмонофосфата, увеличение концентрации которого в клетке замедляет, а уменьшение – ускоряет выделение медиаторов. Антагонистическое действие оказывает циклический гуанозин-монофосфат. Для выделения медиаторов необходимо наличие соответствующего количества двухвалентных катионов кальция и магния.
Имеющиеся экспериментальные данные показывают, что использование ингибиторов простагландинсинтетазы вызывает прекращение выделения жидкости в просвет кишки [Giannella R. A. et al., 1975, 1977]. В связи с тем, что простагландины выступают в желудочно-кишечном тракте не только в качестве медиаторов воспаления, но п участвуют в регуляции интестинальной секреции [Matuchansky С. et al., 1973; Milton-Thompson G. L. et al., 1975], становится понятной их ведущая роль в генезисе диареи при сальмонеллезах. Нельзя забывать и о возможном участии в развитии секреторных расстройств гормонов, вырабатываемых специализированными клетками тонкой кишки, относящимися к APUD-системе, например о вазоактивном интестинальном пептиде [Barbezat G. О., 1973; Schwartz С. J. et al., 1974]. Итак, механизм развития секреторных расстройств при сальмонеллезе в стенке тонкой кишки можно представить себе следующим образом. В результате острого воспаления, возникающего под действием возбудителя, проникающего сквозь слой энтероцитов в собственную пластинку слизистой оболочки, происходит нарушение равновесия между гуморальными системами, осуществляющими регуляцию секреторных процессов в тонкой кишке. Причем основное значение имеет избыток простагландинов. Однако остается неясной роль эндотоксина сальмонелл, который выделяется в этот период в стенке кишки и не может не влиять на течение воспалительного процесса и системы саморегуляции желудочно-кишечного тракта.
Эндотоксин и простагландины
Простагландины представляют собой гидроксилированные продукты превращения полиненасыщенных жирных кислот, состоят из 20 атомов углерода и включают циклопепта повое кольцо. В зависимости от заместителей пятичленного кольца все простагландины делят на девять групп: А, В, С, D, Е, F, G, Н, I, а в зависимости от числа двойных связей в метильной и карбоксильной боковых цепях выделены виды простагландинов в пределах каждой группы: А1, А2, А3, причем цифра указывает на число двойных связей в боковой цепи [Варфоломеев С. Д., Мевх А. Т., 1985]. Исходными продуктами для биосинтеза простагландинов являются ненасыщенные жирные кислоты: дигомогамма-линоленовая, арахидоновая и пентаноевая. Однако наибольшее значение для биосинтеза простагландинов в организме имеет арахидоновая кислота, которая содержится в фосфолипидах клеточных мембран. В липидах тромбоцитов она составляет от 16 до 33% всего содержания жирных кислот. При этом из общего количества кислоты всего 20% обнаружено на внешней стороне плазматической мембраны этих клеток [Schick Р. К. et al., 1981]. Арахидоновая кислота в клетках полностью этерифицирована, на 98% входит в состав фосфолипидов, в свободном состоянии в тканях отсутствует.
Высвобождение арахидоновой кислоты из состава фосфолипидов осуществляется под влиянием особого механизма, действие которого обеспечивает активацию фосфолипазы Аг, ответственной за обеспечение субстратом реакции биосинтеза простагландинов [Irvin R. F., 1982].
После высвобождения из клеточных мембран под действием фосфолипазы А2 она становится субстратом для простагландинсинтетазы. Первоначально из арахидоновой кислоты образуется простагландин Нг, синтез которого обеспечивает эндопероксидпростагландинсинтетаза (циклооксигеназа жирных кислот). Этот процесс не является органоспецифическим и протекает одинаково во всех органах и тканях. Далее синтез простагландинов осуществляется в органах и тканях уже под влиянием других ферментов. Эти ферменты объединены под общим названием простагландин-Н2-конвертаз. В тромбоцитах под действием конвертаз образуется тромбоксан А2, в эндотелиальных клетках сосудов – простациклин (простагландин I). Ферменты везикулярных желез образуют простагландины Е2 и F2, ферменты клеток головного мозга – D2 [Мевх А. Т. и др., 1983]. Прастагландины синтезируются в большинстве органов и тканей, а разрушаются под действием специальной метаболизирующей системы ферментов, которая лучше всего представлена в легких [Марков X. М., 1979].
Простагландины вызывают сильное диуретическое и натрийуретическое действие, влияют на тонус сосудов и уровень артериального давления, моторику желудочно-кишечного тракта, агрегацию тромбоцитов, деятельность центральной и вегетативной нервной системы. Причем простагландины групп Е и F обычно оказывают противоположное действие.
Важное значение имеет открытие высокой биологической активности промежуточных продуктов биосинтеза простагландинов – простагландиновых эндопероксидов, которые оказывают сильное проагрегационное действие на тромбоциты. Исключительно высокие проагрегационные свойства характерны для тромбоксана Аг, он образуется в тромбоцитах из арахидоновой кислоты, а также в почках и оказывает сильное сосудосуживающее действие. При взаимодействии простагландиновых эндопероксидов со структурами сосудистой стенки в эндотелии синтезируется очень активное соединение, получившее название простациклип (простагландин I2) [Moore P. U., Griffiths R. J., 1983]. Особенно важными являются агрегационное и сосудорасширяющее действие простациклина. Поэтому он может служить естественным антагонистом тромбоксана А2. Существует гипотеза о зависимости агрегации тромбоцитов рт количественного соотношения тромбоксана А2 и простациклина [Марков X. М., 1979].
В 1979 г. открыта группа физиологически активных веществ – лейкотриенов, которые синтезируются в цитоплазматических мембранах клеток под влиянием липоксигеназы из арахидоновой кислоты. Лейкотриены образуются в организме повсеместно. Однако основными источниками лейкотриенов являются клетки крови. Наиболее высокое содержание лейкотриенов обнаружено в макрофагах, нейтрофилах и лаброцитах. Лейкотриены участвуют в развитии патологических процессов при заболеваниях, сопровождающихся воспалением и иммунопатологическими процессами [Фролов Е. П. и др., 1984; Piper P. J., 1984].
Механизм действия простагландинов на клетки очень сложен и требует дальнейшего уточнения. Недавно открыты специфические рецепторы к различным группам простагландинов, расположенные на цитоплазматической мембране. Внутриклеточное действие простагландинов, тромбоксанов ж лейкогриенов реализуется с помощью циклических нуклеотидов и ионов кальция. Полагают, что действие производных арахидоновой кислоты на образование циклических нуклеотидов в тканях опосредуется через аденилциклазу. В свою очередь внутриклеточное действие циклических нуклеотидов, например превращение фосфорилазы В в фосфорилазу А, активация протеинкиназ, тесно связано с ионами кальция. Видимо, производные арахидоновой кислоты вызывают деполяризацию и повышение проницаемости; клеточных мембран. При этом внеклеточный кальций устремляется внутрь клетки [Марков X. М., 1979; Фролов Е. П., 1984; Harris В. Н. et al., 1979].
Таким образом, в результате превращения арахидоновой кислоты в организме образуется группа физиологически активных соединений, представленная простагландинамп (ПГ), тромбоксанами (ТХ) и лейкотрйенами (LT) (схема 1).
Схема 1
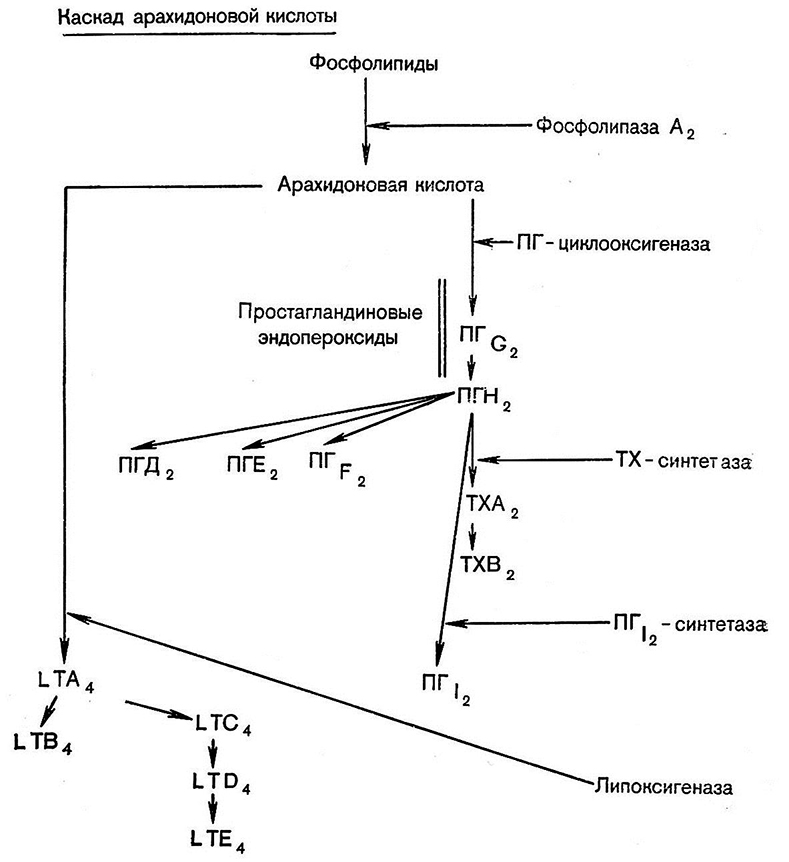
Эти соединения образуются во всех органах и тканях, оказывая на них прямое действие, а также являются посредниками гормонов и других физиологически активных веществ. Это обстоятельство следует учитывать при разработке методов лечения сальмонеллезов. Возможно, действие эндотоксина и на другие системы регуляции гомеостаза реализуется через простагландины. Поэтому представляется целесообразным кратко рассмотреть основные эффекты действия простагландинов, тромбоксанов и лейкотриенов на желудочно-кишечный тракт, системы свертывания крови и регуляции артериального давления, а также учесть их возможное участие в регуляции или реализации действия других физиологически активных веществ – циклических нуклеотидов, ангиотензинов, кининов и катехоламинов.
Простагландины оказывают разнообразное действие на желудочно-кишечный тракт. Они могут как стимулировать, так и тормозить сокращение и секреторную активность тонкой кишки, тормозят секрецию хлористоводородной (НСI) кислоты слизистой оболочки желудка [Wilson D. Е., 1974].
В тонкой кишке представлены главным образом проcтагландины группы Е и F, которые вызывают сокращение продольных мышц тонкой кишки [Bennet A. et al., 1968]. При использовании простагландина E1 для производства аборта в качестве побочных явлений наблюдались боли в животе спастического характера и диарея. Простагландины группы Е вызывают секрецию воды и электролитов в просвет кишки, что клинически проявляется как диарея [Greven J., 1979]. Это подтверждают и результаты опытов по внутривенному введению простагландинов Е2 и F2 добровольцам [Milton-Thompson G. J. et al., 1975]. Простагландины группы Е вызывают также повышение концентрации циклического аденозинмонофосфата в слизистой оболочке тонкой кишки, который выступает, видимо, в качестве посредника при действии простагландинов на энтероциты [Field М. et al., 1981]. В последних синтезируются простагландины, участвующие в регуляции их секреторной активности.
Простагландины образуются в клетках крови (тромбоцитах, лейкоцитах) и сосудистой стенке (простагландин I2). В тромбоцитах, гранулоцитах, лимфоцитах и моноцитах обнаружены ферменты, синтезирующие и метаболизирующие простагландины. Простагландины группы Б влияют на прочность и упругость эритроцитарной мембраны [Гаврилов О. К., Кавешников Б. Ф., 1979; Allen J. Е., Valeri С. R., 1974].
Велика роль производных арахидоновой кислоты, особенно тромбоксанов и простациклина, в процессе свертынания крови. Так, под действием тромбина, АДФ и коллагена, на котором распластывается тромбоцит в процессе образования тромба, усиливается липолиз фосфолипидов мембран. Липолиз вызывает фосфолипаза Аг тромбоцитов. Выделяющаяся в результате липолиза арахидоновая кислота превращается под действием ферментов в циклические эндопероксиды, а затем в тромбоксаны и простациклин. Тромбоксан Аг является основным медиатором местного спазма сосудов и тромбоза. Его антагонистом, ингибитором тромбоза и мощным вазодилататором является простациклин, который синтезируется в микросомах эндотелиальных клеток. Активатором синтеза простациклина является тромбин. Антагонизм в действии тромбоксана Аз и простациклина связан с особенностями механизмов их влияния на аденилатциклазу, определяющую внутриклеточный уровень циклических нуклеотидов. Так, простациклин стимулирует аденилатциклазу, вызывая, таким образом, накопление циклического АМФ в тромбоцитах. Последний подавляет мобилизацию ионов кальция. Тромбоксан Аг, напротив, блокирует аденилатциклазу, обусловливая мобилизацию ионов кальция из тубулярной системы тромбоцитов [Лакин К. М. и др., 1982; Vane J. R., Moncada S., 1980; Weksler В. В., Goldstein I. M., 1982].
Тесно связано с нарушением синтеза простагландинов в организме и развитие диссеминированного внутрисосудистого свертывания (коагулопатия потребления, дефибринация, тромбогеморрагический синдром). Его развитие может быть обусловлено повреждением эндотелиальных клеток, сопровождающееся торможением синтеза простациклина, что вызывает в свою очередь прилипание тромбоцитов к стенке сосудов и их контакту с обнажившимися коллагеновыми волокнами. Это ведет к включению внутреннего пути свертывания крови. Важную роль играет также травматическое повреждение клеток, сопровождающееся выделением тканевой жидкости и освобождением внутриклеточных ферментов [Фермилен Ш., Ферстрате М., 1984]. Учитывая повреждающее действие сальмонеллезного эндотоксина на эндотелиальные клетки кровеносных сосудов микроциркуляториого русла, которые рассматриваются в качестве основной точки приложения токсического действия, можно предполагать, что развитие диссеминированного внутрисосудиетого свертывания при сальмонеллезе связано именно с нарушением синтеза проетациклина и усилением продукции его антагониста тромбоксана Аг [Urbaschek В., Forssinaim W. G., 1980].
Простагландины влияют на деятельность центральной и периферической нервной системы. Существует предположение о разных механизмах действия простагландинов Ег и 1г на адренергическую нейропередачу. P. Hedqvist (1977) полагает, что простагландин Е2 действует на выброс нейропередатчика, т. е. пресинаптически с помощью механизма отрицательной обратной связи, а простагландин 1г влияет на восприятие нейропередатчика, т. е. ностсинаптически, дополняя таким образом действие Простагландин» Е2 ,[Hedqvist Р., 1979].
В ткани мозга экспериментальных животных содержатся в основном простагландины Е1 Е2, F1 и F2, действие которых на уровне нейронов реализуется, видимо, через циклический аденозинмонофосфат [Муратов В. Д., Булаев В. М., 1981]. Простагландины обнаружены практически во всех структурах мозга и спинномозговой жидкости, причем мозг имеет как систему синтеза, так и разрушения простагландинов и тромбоксанов. Простагландины влияют на поведенческие реакции, регуляцию температуры тела, сердечно-сосудистой системы и двигательных функций [Coceani F., 1974; Wolfe L. S., Coceani F. et al., 1979].
Простагландины участвуют в регуляции как местного кровотока, в том числе органного и на уровне микроциркуляториого русла, так и системного артериального давления. Большинство простагландинов оказывает сосудорасширяющее действие. Циркулирующие простагландины и их метаболиты действуют как антигипертензивные гормоны. Действие простагландинов, которые синтезируются почками, реализуется путем сложного взаимодействия с ренин-ангиотензиновой и калликреин-кининовой системами органа. Стенки артерий и вен млекопитающих продуцируют значительные количества простагландинов Е2 и F2, которые участвуют, видимо, в регуляции местного кровообращения.
Основными циркулирующими простагландинами являются простагландины группы А. Они в отличие от групп Е и F не разрушаются в легких. Однако наибольшее значение имеет простациклин, который не только не метаболизм руется в легких, но и способен достигать периферических сосудов, где реализуется его действие. При внутрипенном введении простагландинов Е, А или I2 артериальное давление снижается за счет расширения периферических сосудов – артериол, прекапилляров и венул. Простагландин F2 обычно вызывает незначительное повышение артериального давления. Снижение артериального давления, вызванное простагландинами А и Е, кратковременное и сопровождается компенсаторным увеличением частоты сердечных сокращений и сердечного выброса. Простагландин Е предотвращает спазм сосудов, развивающийся под действием катехоламинов, ангиотензина II и антидиуретического гормона. Действие простагландинов устраняется ингибиторами простагландинсинтетазы, например индометацином [Heinemann Н. О., Lee J. В., 1976; Saynavalammi P. et al., 1979].
Особое место в системе регуляции артериального давления занимает почка. Простагландинподобные соединения в ней открыты J. В. Lee и соавт. в 1965 г. Позже было установлено, что в почке синтезируются главным образом простагландины А2, Е2 и F2, а также тромбоксаны А2 и В2. В эндотелии сосудов образуется простациклин. Основным 1 фостагландином почки в количественном отношении является простагландин Е2, синтез которого осуществляется преимущественно в нефроцитах собирательных трубок и иптерстициальных клетках мозгового вещества {Muirliead Е. Е. et al., 1972; Bohman S–О., 1977]. Простагландины оказывают на почку преимущественно локально© действие, так как они быстро поступают в венозную кровь или мочу либо разрушаются. В корковом веществе почки содержится фермент 15-гидроксипростагландиндегидрогепаза I типа, которая инактивирует простагландины.
Простагландины в почке оказывают различное физиологическое и фармакологическое действие: вызывают сильный диуретический и натрийуретический эффект, обладают мощным влиянием на тонус сосудов, уровень артериального давления, агрегацию тромбоцитов, деятельность вегетативной нервной системы, секрецию гормонов, регулируют действие последних на «ткани-мишени». Видимо, простагландины почек играют определяющую роль в механизмах регуляции гомеостаза, в обеспечении которых ночка как орган выделения занимает одно из центральных мест.
Предположения об антигипертензивном действии мозгового вещества почки убедительно подтвердились после экспериментов с пересадкой мозгового вещества животным с экспериментальной гипертензией и достижением таким образом гипотензивного эффекта. В связи с этим важное значение имеет прямая зависимость концентрации простагландина Е2 от активности ренина в плазме крови [Attalah A. A. et al., 1982]. Действие простагландинов, возбуждающее синтез ренина, реализуется, видимо, через основные механизмы, обеспечивающие регуляцию выброса ренина, – через барорецепторы, плотное пятно или путем прямого воздействия на юкстагломерулярные клетки [Gerber J. G., Nics A. S., 1981; Henrich W. L., 1981]. Наиболее мощное влияние на синтез ренина оказывает простациклин, который синтезируется эндотелием приносящей артериолы и, вероятно, является основным простагландином в корковом слое почки [Whorton A. R. et al., 1978]. Это доказывают многочисленные эксперименты [Марков X. М. и др., 1980; Frolich J. С, Fojes-Toth G., 1982]. Стимуляторы синтеза ренина простагландины Е2 и I2 вызывают снижение концентрации циклического аденозинмонофосфата в тканях. Поэтому вторичным передатчиком стимулирующего действия простагландинов на синтез ренина вполне может быть циклический аденозинмонофосфат [Okahara Т. et al., 1977; Campbell W. В. et al., 1979].
Под действием простагландинов, в основном Е2, параллельно с увеличением почечного кровотока возрастает натрий- и диурез. Простагландин Е2 снижает реабсорбцию натрия в дистальном отделе нефрона [Fulgraff G., Meiforth А., 1971]. Его действие реализуется на уровне толстого сегмента восходящего отдела петли нефрона JHigashihara E. et al., 1979]. Усиление диуреза связано с нарушением равновесия между выделением антидиуретического гормона и простагландином [Kirschenbaum, М. A. et al., 1982].
Действие простагландинов на почечный кровоток, натрий- и диурез реализуется при непосредственном участии калликреин-кининовой системы. По-видимому, простагландины группы E, особенно Е2, являются посредниками при действии кининов на клетки и ткани. Под влиянием брадикинина, например в почках, повышается концентрация простагландинов Е2 и I2 [Kauker М. L., 1980].
Простагландины обладают также некоторыми свойствами медиаторов иммунного ответа, особенно простагландины E1, Е2 и F2α [Медуницын Н. В. и др., 1980]. Так, простагландины E1 и Е2 являются сильными ингибиторами ответа Т-лимфоцитов. Торможение синтеза простагландинов усиливает иммунный ответ. Простагландины влияют на способность В-лимфоцитов продуцировать антитела, т. е. участвуют в регуляции гуморального иммунного отпета [Goodwin J. S., Webb D. R., 1980]. Действие простагландинов реализуется на ранних стадиях активации лимфоцитов и макрофагов [Webb D. R. et al., 1979]. Полиморфно-ядерные лейкоциты способны к выработке тромбоксанов в ответ на стимуляцию [Weksler В. В., Goldstein J. М., 1980].
Простагландины реализуют свое действие в лимфоцитах через циклический аденозинмонофосфат в присутствии ионов кальция. Причем антагонистом циклического аденозинмонофосфата может выступать гуанозинмонофосфат. Доказано участие циклических нуклеотидов в регуляции дифференцировки лимфоцитов, пролиферации тимоцитов и продукции антител [Hadden J. W. et al., 1979].
Таким образом, биологическое действие простагландинов, тромбоксанов и лейкотриенов чрезвычайно разнообразно. Наряду с участием в регуляции гладкой мускулатуры, системы свертывания крови, инкреторной и экскреторной функций почек, иммунной системы они являются модуляторами действия различных гормонов. Необычайно широкий спектр действия простагландинов, тромбоксанов и лейкотриенов связывают с тем, что они влияют на образование в клетках циклического аденозинмонофосфата – универсального регулятора внутриклеточного гомеостаза. При этом важную роль играет и концентрация нонов кальция в «клетке-мишени». У больных сальмонеллезом между концентрациями циклического аденозинмонофосфата в энтероцитах и кальция в плазме крови установлена обратная связь. Причем уровень этого циклического нуклеотида находится в прямой связи с показателями, характеризующими изменения водно-электролитного гомеостаза [Рябов В. И., 1982]. В связи с тем что циклические нуклеотиды (аденозин- и гуанозинмонофосфат) являются по существу универсальными внутриклеточными регуляторами, необходимо рассмотреть их биологическую роль в организме. Полагают, что и действие спльмонеллезного эндотоксина на уровне клеток может реализоваться через систему циклических нуклеотидов [Вунип К. В. и др., 1977].
Превращение аденозинтрифосфата в циклический аденозинмонофосфат происходит при участии фермента аденилатциклазы в присутствии ионов кальция и магния. Гидролиз циклических нуклеотидов с образованием нециклических нуклеозидмонофосфатов катализируют фосфодиэстеразы. Активаторами аденилатциклазы являются различные гормоны и физиологически активные вещества. Циклические нуклеотиды активируют протеинкиназы, обеспечивающие перенос остатков фосфорной кислоты на определенные аминокислоты, что и ведет в конечном итоге к изменению функции клетки. Так в общих чертах можно представить себе роль циклических нуклеотидов в механизмах внутриклеточной регуляции. Наряду с возможным участием циклических нуклеотидов в реализации действия эндотоксина на уровне клетки и их посреднической ролью для простагландинов, тромбоксанов и лейкотриенов необходимо упомянуть о значении циклических нуклеотидов в системе гемостаза и иммунорегуляторных процессах. Накопление кальция в тромбоцитах вызывает прилипание, агрегацию и ретракцию последних. В то же время стимуляция аденилатциклазы или торможение фосфодиэстеразы приводит к усилению образования циклического аденозинмонофосфата и одновременно удалению кальция из клетки [Федоров Н. А., 1979]. Изменение уровня циклических нуклеотидов является также модулятором различных реакций иммунокомпетентных клеток. Содержание циклического аденозинмонофосфата в иммунокомпетентных клетках отражает их зрелость, меняется при различных заболеваниях, затрагивающих иммунную систему организма [Бирюков А. В. и др., 1985].
Нами изучены молекулярные основы патогенеза сальмонеллезной интоксикации на основе оценки роли простагландинов в ответной реакции макроорганизма на введение сальмонеллезного эндотоксина. В процессе работы проведен анализ влияния эндотоксина сальмонелл на течение реакции биосинтеза простагландинов из арахидоновой кислоты в условиях in vitro, а также исследована роль действия препаратов, подавляющих биосинтез простагландинов при сальмонеллезной интоксикации. Использована методика биосинтеза простагландинов in vitro, имитирующая условия биосинтеза, складывающиеся при сальмонеллезе, острый период которого характеризуется выраженной эндотоксинемией. В качестве предшественника простагландинов использована арахидоновая кислота высокой степени частоты (91-99%), выделенная из липидов семенников и поджелудочной железы животных по мотоду Р. В. Бобылева и соавт. (1978). Источником простпгландинсинтетазы были пузырьковые железы баранов. Фермент приготовляли в виде супернатанта (Ажгихин И. С., 1978]. Постановка реакции в стандартном варианте, предложенном Р. В. Бобылевым и соавт. (1980), являлась постоянным контролем для конкретных образцов арахидоновой кислоты и нростагландинеинтетазы, используемых в параллельных опытах биосинтеза. В первой серии опытов в реакцию биосинтеза простагландинов вводили в качестве дополнительного компонента эндотоксин S. typhimurium и S. typhi. Указанные, а также все другие использованные нами эндотоксины были получены методом Буавена. Во всех опытах 100 мг натриевой соли арахидоновой кислоты эмульгировали в 50 мл фосфатного буфера рН 8,0, добавляли 25 мг глутатиона и 400 мг гидрохинона. 50 мг эндотоксина растворяли в 60 мл фосфатного буфера и смешивали с супернатантом пузырьковых желез барана. Оба раствора раздельно термостатировали при температуре 37,0±9,5°С, затем объединяли и пропускали воздух в течение 20 мин. Реакцию биосинтеза прерывали путем добавления 1,8 л этилового спирта и оставляли на 2 ч при температуре 4°С, затем центрифугировали. Супернатант сливали и упаривали при температуре бани 45 ± 1 °С в вакууме до конечной концентрации спирта в 66 °С. Остаток экстрагировали петролейным эфиром, а водно-спиртовой осадок вновь упаривали до полного отсутствия спирта, Водную фазу подкисляли до рН 3,5 и экстрагировали диэтиловым эфиром. Эфирный экстракт промывали равным объемом дистиллированной воды, сушили над Na2SO4 и упаривали досуха. Сухой остаток растворяли в этиловом спирте и производили анализ на выявление структуры и свойств веществ с помощью спектрофотометрических и других методов.
При дополнительном введении эндотоксина S. typhimurmm в реакцию биосинтеза простагландинов из арахидоновой Кислоты (табл. 4) в контрольном опыте выход простагландинов составил 8,0±0,7 мг, а при добавлении эндотоксина – 25,6±1,2 мг (Р<0,001).
Таблица 4.
Биосинтез простагландинов in vitro из арахидоновой кислоты
при дополнительном введении эндотоксина

Таким образом, в модельном опыте выход простагландинов более чем в 3 раза превысил результаты контрольного опыта. При внесении в реакцию биосинтеза эндотоксина S. typhi выход простагландинов составил 28,0±0,3 мг. Оба эндотоксина более чем в 3 раза усиливали синтез простагландинов из арахидоновой кислоты.
Сопоставление результатов контрольного и модельного опытов позволило получить принципиально новые представления о свойствах эндотоксина сальмонелл, заключающихся в способности резко усиливать биосинтез простагландинов из арахидоновой кислоты, широко представленной во всех тканях организма человека и животных. Практически идентичность процессов биосинтеза в условиях in vivo и in vitro позволяет считать, что в условиях ондотоксинемии, когда в организме больного человека или экспериментального животного имеется большое количество эндотоксина (острый период заболевания или парентеральное введение эндотоксина), процессы биосинтеза резко усиливаются, возникает избыточное количество этих тканевых гормонов, что не может не вызвать изменений функционального состояния различных систем организма.
Во второй серии опытов нами была предпринята попытка выяснения механизмов усиления синтеза простагландинов, наступающего под влиянием эндотоксина. С этой целью из реакций биосинтеза был исключен мультиферментный комплекс простагландинсинтетазы при сохранении арахидоновой кислоты и других указанных выше ингредиентов реакции. Арахидоновую кислоту и эндотоксин сальмонелл смешивали в соотношении 1:7. В опытах были использованы эндотоксины S. typhimurmm, S. newport, S. typhi, E. coli. Во всех случаях методами спектрофотометрии, масс-спектрометрии, биотестирования, тонкослойной хроматографии обнаружены простагландины различных классов. Количественные соотношения ПГЕ2 ПГА2 и ПГF2, полученные под влиянием эндотоксина S. typhimurium, составили 2:1: следы. Соответственно для эндотоксина S. newport, Е. coli, S. typhi эти соотношения имели значения 10:5:1:1:1:0:1:1:0.
В контрольных опытах (арахидоновая кислота + прогтагландинсинтетаза) были выявлены простагландины Е, А и F2α в соотношении 12:5:1. Следует подчеркнуть, что количество эндотоксина, использованного в этой серии опытов (700 мг), значительно превышало его уровни в первой серии опытов (50 мг). Причины трансформации арахидоновой кислоты в простагландины под влиянием эндотоксина остаются не вполне ясными, что требует дополнительного изучения. Таким образом, исследование нзаимодействия эндотоксина сальмонелл Е. coli на течение реакций биосинтеза простагландинов из арахидоновой кислоты позволило установить их прямое стимулирующее влияние на синтез. Выявлено два возможных пути реализации этого явления:
а) стимуляция мультиферментного комплекса простагландинсинтетазы;
б) усиление автокатализа арахидоновой кислоты в простагландины.
Принимая во внимание результаты опытов in vitro и их идентичность биосинтезу простагландинов в условиях in vivo, нами проведено изучение стимулирующего влияния эндотоксина S. typhimurium на биосинтез простагландинов из эндогенной арахидоновой кислоты у кроликов породы Шиншилла. При этом мы исходили из предположения, что усиление синтеза простагландинов, наступающее под влиянием эндотоксина, является основой функциональных нарушений. В качестве тестов для изучения патогенетической роли простагландинов у животных оценивали состояние гемостаза, степень выраженности диареи, частоту дыхания и сердцебиения, ректальную температуру, воспроизводимость местной и генерализованной реакций Шварцмана. Во всех трех сериях опытов животным вводили индометацин: перед инъекцией эндотоксина (за 1 ч) и через 1-1 1/2 ч после инъекций. Индометацин вводили однократно через зонд в желудок животного в дозе 10 мг/кг.
ЛД50 серии эндотоксина S. typhimurium, использованного в экспериментах, составила 1,89 мг/кг массы тела. При однократной инъекции эндотоксина в дозе 1 мг/кг у животных развивались диарея, адинамия, учащение сердцебиений, одышка. Потеря массы тела к исходу суток достигала 8-10% от начальной (табл.5).
Таблица 5.
Характеристика проявлений биологической активности эндотоксина
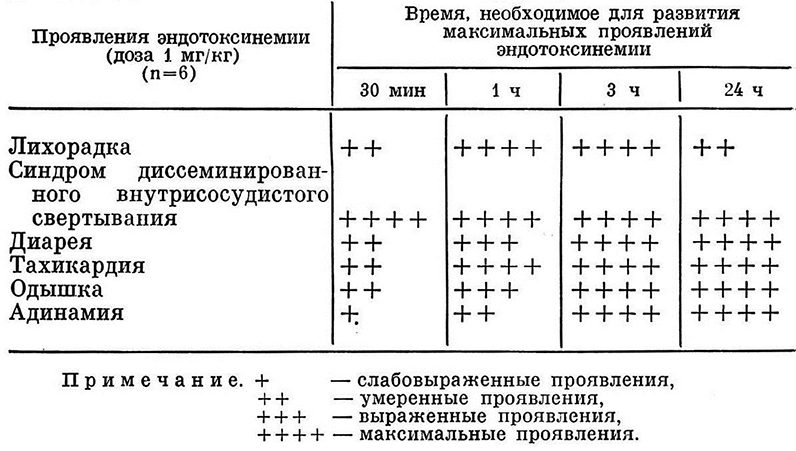
При исследовании гемостаза через 30 мин после инъекции эндотоксина отмечены признаки выраженной гипокоагуляции. Максимальные проявления гипокоагуляции выявлены через 24 ч от начала опытов. При исследовании коагулограммы отмечено достоверное увеличение фибринолитической активности, времени рекальцификации и свободного гепарина, тромбинового времени, уровней растворимых комплексов фибринмономеров (Р<0,05 во всех случаях). Показатели концентрации фибриногена, количества тромбоцитов были достоверно снижены (Р<0,05).
Тромбоэластографическое исследование в целом отражало динамику коагулографических изменений. Уже через 30 мин отмечено достоверное удлинение времени реакции, времени образования сгустка, специфической константы коагуляции, показателей синерезиса и тотального времени свертывания крови. Угловая константа и индекс коагуляции значительно уменьшались (Р<0,05). При повторных исследованиях через 3 и 24 ч отмечено прогрессирование гипокоагуляционных изменений.
Одновременно выявлены значительные нарушения микроциркуляции в почках, представленные расширением клубочковых и перитубулярных капилляров, агрегацией в них эритроцитов, появлением полиморфно-ядерных лейкоцитов. В просвете капилляров клубочка появлялись нити фибрина. Описанные изменения были особенно значительными через 24 ч после введения экспериментальным животных сальмонеллезного эндотоксина (рис. 1).
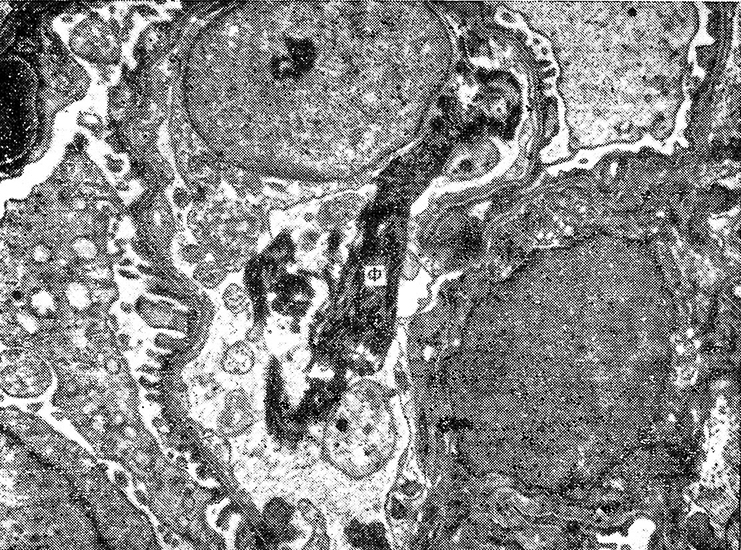
Рис. 1. Введение садьмонеллезиого эндотоксина (24 ч опыта).
Фибрин (Ф) в просвете капилляра почечного клубочка. X 10000.
При этом в строме ворсинок и подслизистом слое тонкой кишки обнаружены резкое расширение сосудов микро-циркуляторного русла и отек стромы, в инфильтрате увеличивалось количество плазматических клеток (рис. 2).
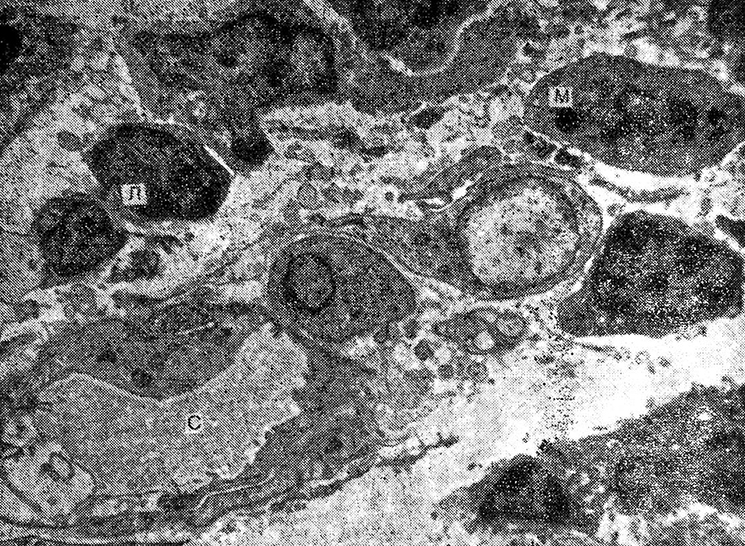
Рис. 2. Введение сальмонеллезного эндотоксина (24 ч опыта).
Расширение сосудов (С) и отек стромы ворсинок тонкой кишки,
лимфоидные клетки (Л) и макрофаги (М) в составе инфильтрата: х 6000.
При воспроизведении местной и генерализованной реакций Шварцмана инъекция 2-й дозы эндотоксина в обоих случаях сопровождалась резкими гипокоагуляционными изменениями коагулограммы и тромбоэластограммы. Через 2-3 ч после инъекции разрешающей дозы при воспроизведении местной реакции Шварцмана в месте инъекции «подготовительной» дозы развивались характерные некротические и геморрагические изменения. При воспроизведении генерализованной реакции Шварцмана инъекция разрешающей дозы вызывала гибель 58% животных в течение 4-6 ч.
При воспроизведении местной реакции Шварцмана лиутрикожное введение эндотоксина уже через 30 мин сопровождалось увеличением свертывающего потенциала крови. Тромбиновое (и в меньшей степени протромбиновое) время уменьшалось (Р<0,02), уровни РКФМ плазмы крови увеличивались (Р<0,001). Количество тромбоцитом уменьшилось с 354,8±5,1 до 338,7± 11,3 тыс. в 1 мм3 (Р<0,05). Показатели тромбоэластограммы отражали тенденцию к повышению свертывающего потенциала крови (рис. 3).
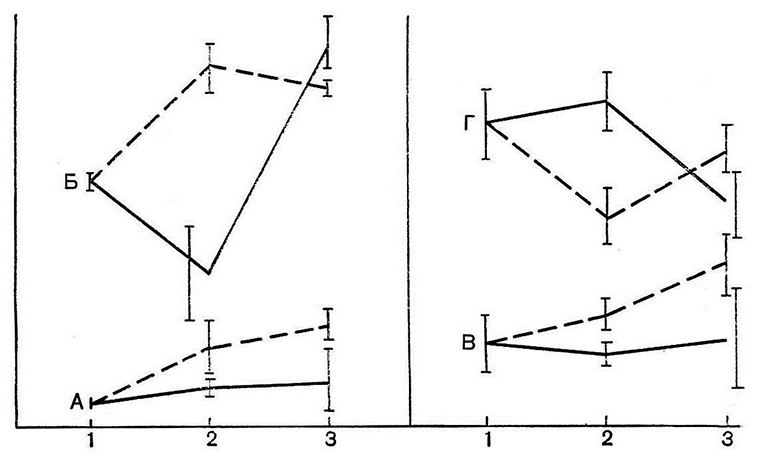
Рис. 3. Состояние гемостаза у животных при воспроизведении местной реакции Шварцмана.
А – растворимые комплексы фибрин-мономеров (РКФМ),
Б – концентрация фибриногена плазмы, В – протромбиновое время, Г – тромбиновое время;
1 – исходные данные, 2 – через 3 ч после инъекции 1-й дозы эндотоксина,
3 – через 3 ч после инъекции 2-й дозы эндотоксина.
Сплошная линия – влияние индометацина на гемостаз; пунктирная –
динамика показателей в контрольной группе животных.
Внутривенное введение «разрешающей» дозы эндотоксина существенно изменяло параметры гемостаза. Показатели протромбинового времени, концентрации фибриногена, РКФМ увеличивались (Р<0,05), а количество тромбоцитов было достоверно ниже исходных значений. Большинство параметров тромбоэластограммы были характерными для состояния гипокоагуляции.
При воспроизведении генерализованной реакции Шварцмана внутривенная инъекция первой дозы эндотоксина сопровождалась повышением свертывающего потенциала крови с одновременной активацией фибринолиза (повышение уровня РКФМ). Уменьшалось количество тромбоцитов (Р<0,05) (рис. 4). Большинство параметров тромбоэластограммы отражало состояние гиперкоагуляции.
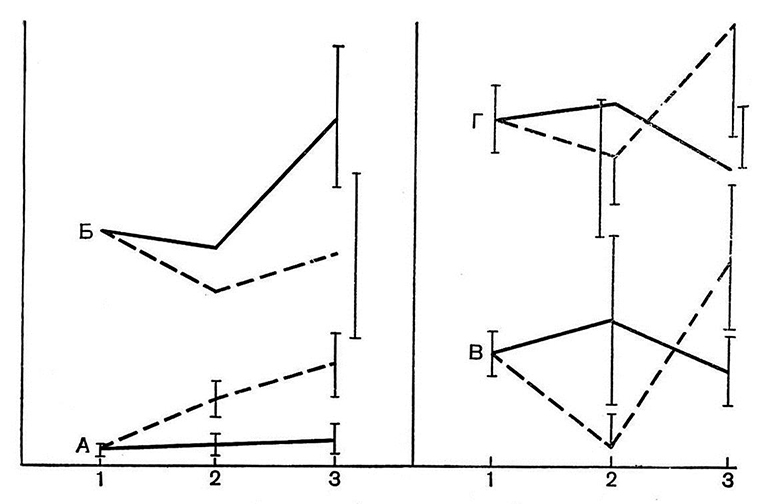
Рис. 4. Состояние гемостаза у животных
при воспроизведении генерализованной реакции Шварцмана.
Условные обозначения те же, что и на рис. 3.
Внутривенное введение 2-й дозы эндотоксина приводило к гипокоагуляционным изменениям. Через 30 мин после инъекции 2-й дозы эндотоксина тромбиновое и протромбииовое время увеличивалось, а количество тромбоцитов значительно уменьшалось (291,3 ±6,9 тыс., Р<0,05). Концентрация фибриногена и уровни РКФМ были выше исходных значений. Состояние гипокоагуляции отражали и показатели тромбоэластограммы. Исследование показателей гемостаза, проведенное через 3 ч после инъекции 2-й дозы эндотоксина, выявило продолжающиеся гипокоагуляционные изменения коагулограммы и тромбоэластограммы. Через сутки после инъекции эндотоксина явления гипокоагуляции еще оставались.
Исследования гемостаза при местной и генерализованной реакциях Шварцмана выявили способность эндотоксина вызывать синдром диссеминированного внутрисосудистого свертывания, который наблюдался и при однократном введении эндотоксина. Установлено, что в отличие от однократного введения эндотоксина при местной и генерализованной реакциях Шварцмана инъекция первой дозы эндотоксина сопровождалась тенденцией к повышению свертывающего потенциала крови. Не исключено, что при большой дозе эндотоксина (1,0 мг/кг), использованной в первой серии опытов, фаза гиперкоагуляции чрезвычайно скоротечна и, наоборот, подкожная и внутривенная инъекции малых доз эндотоксина (соответственно местная и генерализованная реакции Шварцмана) приводят к замедленному развитию фазы гиперкоагуляции.
Влияние эндотоксина на обмен фибриногена достаточно сложное. При однократном введении эндотоксина в дозе 1,0 мг/кг выявлены низкие концентрации фибриногена, а при воспроизведении местной и генерализованной реакций Шварцмана даже после введения второй дозы эндотоксина имела место тенденция к увеличению концентрации фибриногена.
Анализ состояния гемостаза у животных с местной и генерализованной реакциями Шварцмана после инъекции «подготовительной» дозы эндотоксина (0,1 мг/кг) и при его однократном введении дает возможность объяснить разницу в параметрах гемостаза у больных с легким течением сальмонеллеза, с одной стороны, тяжелым и среднетяжелым – с другой. Мы полагаем, что тенденция к повышению свертывающего потенциала крови, чаще отмечаемая у больных с легкой степенью тяжести, определяется меньшей степенью эндотоксинемии, наблюдаемой у этих больных. Об этом свидетельствуют данные И. В. Рубцова и М. X. Галимзянова (1979) о том, что при тяжелом и среднетяжелом течении заболевания в крови больных концентрация эндотоксина выше, чем при легком.
Таким образом, закономерное чередование фаз синдрома диссеминированного внутрисосудистого свертывания крови происходит как при однократной инъекции эндотоксина, так и при воспроизведении местной и генерализованной реакций Шварцмана.
Большая длительность гиперкоагуляционной фазы при феноменах Шварцмана определяется дозой эндотоксина, которая в условиях нашего эксперимента была в 10 раз меньше, чем при однократном его введении.
При воспроизведении генерализованной и местной реакций Шварцмана введение первой дозы эндотоксина (0,1 мг/кг) не вызывает существенных нарушений микроциркуляции и повреждения внутренних органов.
В почках животных с местной реакцией Шварцмана, набитых после введения 2-й дозы эндотоксина, в просвете капилляров клубочка найдены скопления фибрина (рис. 5).
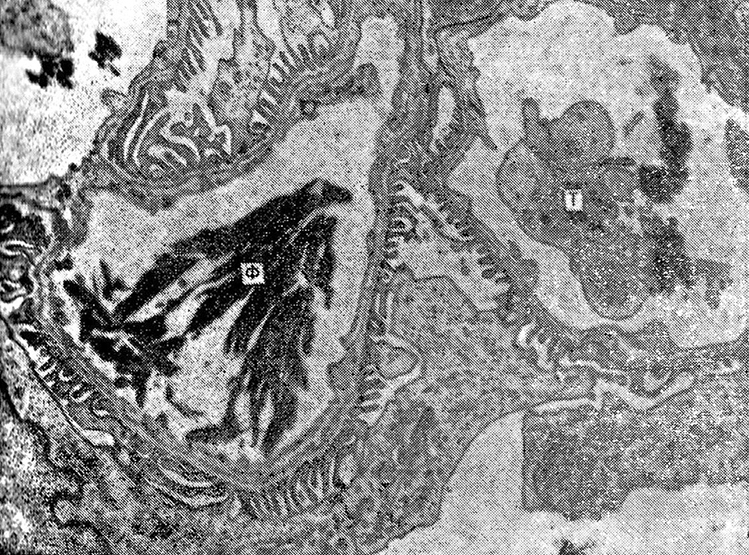
Рис. 5. Местная реакция Шварцмана. Второе введение эндотоксина.
Фибрин (Ф) в просвете капилляров почечного клубочка; Т – тромбоциты. х 10 000.
Перитубулярные капилляры резко расширены, в их просвете – форменные элементы крови. Цитоплазма нифроцнтов канальцев набухшая, вакуолизированная, митохондрии набухшие, с редуцированными кристами. Мозговое вещество отечное. Цитоплазма интерстициальных клеток мозгового вещества вакуолизирована.
При генерализованной реакции Шварцмана в клубочках почек выявлены выраженная вакуолизация и набухание эндотелия, появление фибрина и полиморфно-ядерных лейкоцитов с секвестрированными лизосомами (рис. 6).
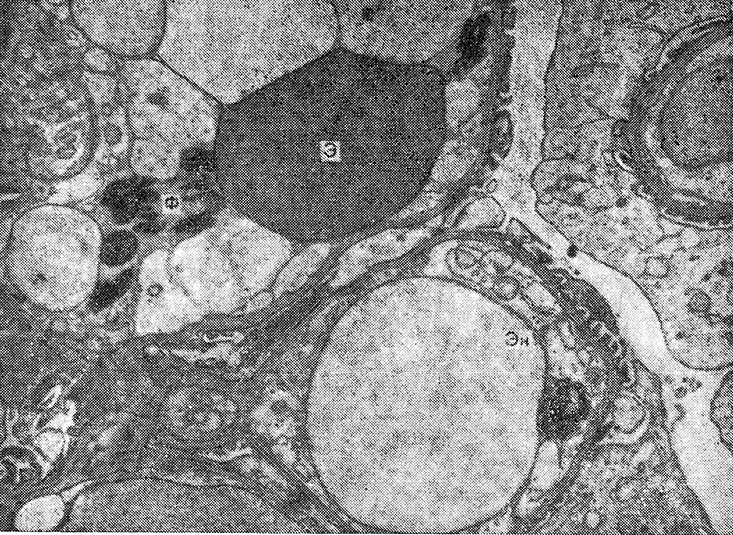
Рис. 6. Генерализованная реакция Шварцмана. Второе введение эндотоксина.
Выраженная вакуолизация эндотелия (Эн) капилляров почечных клубочков,
эритроцит (Э) и фибрин (Ф) в просвете капилляров. X 10 000.
Пертубулярные капилляры резко расширены, эндотелий в них набухший, вакуолизированный, в просвете – скопление форменных элементов крови (рис. 7).
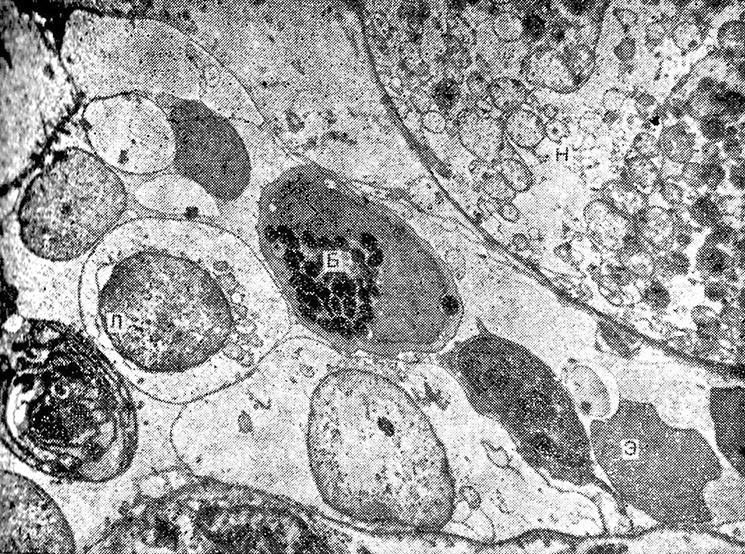
Рис. 7. Генерализованная реакция Шварцмана. Второе введение эндотоксина.
Резкое расширение перитубулярного капилляра почки. Просвет капилляра заполнен форменными элементами крови:
притроцитами (Э), лимфоцитами (Л), базофилами (Б). Цитоплазма нефроцита (Н) вакуодизирована. х 6000.
Отмечены выраженный отек мозгового вещества и вакуолизация его интерстициальных клеток (рис. 8).
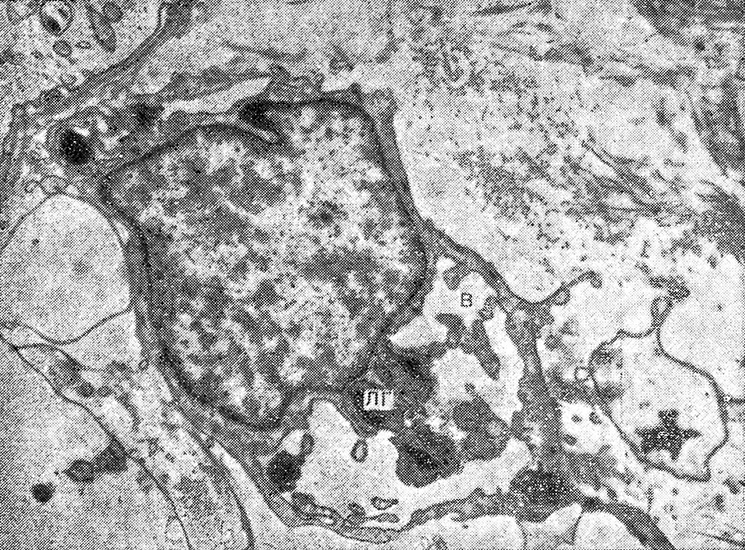
Рис. 8. Генерализованная реакция Шварцмана. Второе введение эндотоксина.
Отек интерстиция мозгового вещества почки, вакуолизация цитоплазмы (В) интерстициальной клетки;
ЛГ – липидные гранулы. х 15 000.
Таким образом, результаты изучения действия эндотоксина при одно- и двукратном его введении (местная и генерализованная реакция Шварцмана) выявили в целом однонаправленные изменения гемостаза и нарушения в системе микроциркуляции. Последние характеризовались выпадением фибрина в микроциркуляторном русле исследованных органов, особенно почек, и повреждением в последних перитубулярных капилляров.
Механизм влияния эндотоксина на состояние гемостаза и микроциркуляции сложен и не до конца ясен. Во многих исследованиях было показано, что эндотоксин грамотрицательных бактерий способен резко изменить функциональное состояние тромбоцитов, вызывать их агрегацию. Последнее является существенным фактором, ведущим к развитию тромбогеморрагических осложнений.
Известно, что активированные тромбоциты способны синтезировать простагландины из арахидоновой кислоты, входящей в состав фосфолипидов мембран [Smith I. В., Silver М. Т., 1976]. Добавление арахидоновой кислоты к плазме, богатой тромбоцитами, вызывает агрегацию последних. Одновременно возрастает синтез простагландинов. Все промежуточные продукты простагландинов синтеза стимулируют (простагландины С2 и Н2, тромбоксап А2) или угнетают (простагландины E1, D2) агрегацию тромбоцитов [Ажгихин И. С., 1978; Гаврилов О. К., 1981].
Нами проведено изучение влияния предшественника простагландинов – арахидоновой кислоты – на агрегацию донорских тромбоцитов. В качестве индукторов агрегации использованы АДФ (1:103 мкг/мл), эндотокенп S. typhimurium, полученный по методу Буавена (1:10 мкг/мл), и натриевая соль арахидоновой кислоты (50 мкг/кг). Проведено две серии опытов. В первой серии (n=36) в качестве индуктора агрегации использован АДФ. При этом к образцам богатой тромбоцитами плазмы, помещенным в прибор, последовательно добавлялись по 0,1 мл эндотоксина и натриевой соли арахидоновой кислоты. Во второй серии опытов (n=36) образцы богатой тромбоцитами плазмы предварительно инкубировали (60 мин) с эндотоксином и солью арахидоновой кислоты, взятыми в тех же объемах и концентрациях. Затем образцы богатой тромбоцитами плазмы помещали в агрегометр и определяли способность тромбоцитов к агрегации при добавлении АДФ. В обеих сериях опытов объем образца богатой тромбоцитами плазмы составлял 0,7 мл.
Первая серия опытов позволила оценить способность как эндотоксина, так и арахидоновой кислоты индуцировать агрегацию тромбоцитов. При непрерывной регистрации процесса агрегации тромбоцитов обнаружено, что добавление к образцам богатой тромбоцитами плазмы арахидоновой кислоты индуцировало агрегацию тромбоцитов. Суммарная степень агрегации тромбоцитов при последующем добавлении к этим образцам АДФ была выше, чем при действии только АДФ.
Добавление к образцам богатой тромбоцитами плазмы эндотоксина также индуцировало агрегацию тромбоцитов. Степень агрегации колебалась от 2 до 15 мм. Последующее добавление к этим же образцам АДФ вызывало усиление агрегации тромбоцитов.
Действие эндотоксина на агрегацию тромбоцитов было более выраженным при его введении после арахидоновой кислоты. Суммарная степень агрегации тромбоцитов при последовательном добавлении к богатой тромбоцитами плазме арахидоновой кислоты, эндотоксина и АДФ была отчетливо выше контрольной.
Таким образом, присутствие арахидоновой кислоты в образцах богатой тромбоцитами плазмы стимулировало агрегацию тромбоцитов, вызываемую как АДФ, так и эндотоксином.
Во второй серии опытов выявлены несколько иные закономерности. Длительное инкубирование образцов богатой тромбоцитами плазмы с эндотоксином приводило к отчетливому снижению степени агрегации тромбоцитов, что связано с длительным воздействием эндотоксина на тромбоциты и повреждением последних. Об этом свидетельствует выпадение части тромбоцитов в виде агрегатов.
Длительная инкубация тромбоцитов с эндотоксином в присутствии арахидоновой кислоты приводила к более высокой степени агрегации под влиянием последующего инодепия АДФ. Следовательно, как и в предыдущей серии опытов, присутствие арахидоновой кислоты даже при длительном воздействии на тромбоциты эндотоксином оказывало стимулирующее влияние на проявление агрегациоиных свойств тромбоцитов. Механизм такого воздействия арахидоновой кислоты на реакцию агрегации тромбоцитов, вероятно, связан с вовлечением этой кислоты в биосинтез.
Таким образом, исследование действия эндотоксина сальмонелл Е. coli на течение реакции биосинтеза простагландинов из арахидоновой кислоты позволило установить их прямое стимулирующее влияние на синтез. Выпилено два возможных пути реализации этого явления:
а) стимуляция мультиферментного комплекса простагландинсинтетазы;
б) усиление автокатализа арахидоновой кислоты в простагландины.
Принимая во внимание результаты опытов in vitro, которыо достоверно воспроизводят биосинтез простагландинов в условиях in vivo, нами проведено изучение стимулирующего влияния эндотоксина S. typhimurium на биосинтез простагландинов из эндогенной арахидоновой кислоты у кроликов породы Шиншилла. При этом мы исходили из предположения, что усиление синтеза простагландинов, происходящее под влиянием эндотоксина, является основой тех функциональных нарушений, которые были описаны ранее в экспериментальных работах. В качестве тестов для изучения патогенетической роли простагландинов у животных оценивали состояние гемостаза, степень водно-электролитных потерь, частоту дыхания и сердцебиения, ректальную температуру, воспроизводимость местной и генерализованной реакцией Шварцмана. В качестве ингибитора биосинтеза простагландинов использован индометацин.
Во всех трех сериях опытов (однократная внутривенная инъекция эндотоксина в дозе 1 мг/кг, воспроизведение местной и генерализованной реакцией Шварцмана) индометацин вводили однократно в дозе 10 мг/кг. При этом 50% животных в каждой серии опытов препарат вводили за 1 ч до инъекции эндотоксина, а остальным – через 1-3 ч после инъекции эндотоксина.
В предварительных опытах нами не выявлено существенных изменений в показателях коагулограммы и тромбоэластограммы после введения индометацина в дозе 10 мг/кг. Некоторое снижение свертывающего потенциала крови мы объясняем влиянием индометацина на агрегационную способность тромбоцитов. Известно, что торможение синтеза простагландинов тромбоцитами существенно снижает агрегационные свойства последних.
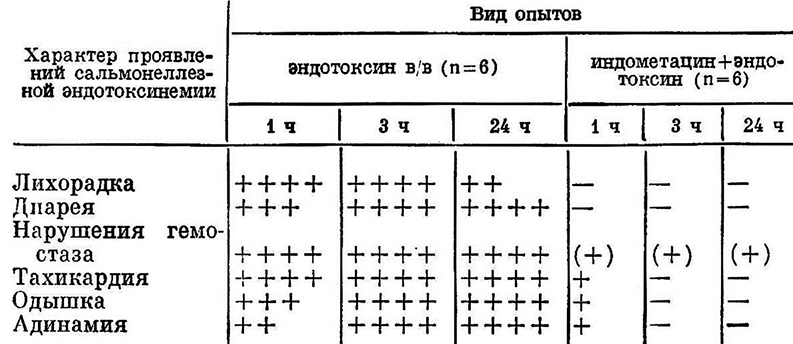
Инъекция эндотоксина после введения ипдометацина не сопровождалась развитием характерных признаков сальмонеллезной эндотоксинемии (табл. 6).
Таблица 6.
Характеристика влияния предварительного введения индометацииа
на развитие симптомов эндотокеинемии у кроликов
Примечание. Условные обозначения: см. табл. 5.
(+) – непостоянный признак.
Резкое снижение выраженности клинических проявлений сальмонеллезной эндотоксинемии сопровождалось у животных отсутствием отчетливых признаков синдрома диссеминировэнного внутриеосудистого свертывания крови, за исключением повышения уровней РКФМ.
Способность индометацина предотвращать развитие как клинических проявлений сальмонеллезной интоксикации, так и нарушений в системе гемостаза при его предварительном введении свидетельствует о том, что генезис этих нарушений связан с усилением синтеза простагландинов под влиянием эндотоксина. Значительный интерес представляет изучение эффективности индометацина при развившейся интоксикации (табл. 7). После введения индометацииа клинические проявления эндотокеинемии у животных быстро купировались.
Таблица 7.
Характеристика клинических проявлений эндотокеинемии
при введении индометацииа после инъекции эндотоксина у кроликов

Примечание. Условные обозначения: см. табл. 5.
При анализе коагулограмм установлено, что лишь уровни РКФМ превышали исходные значения (Р<0,001). Индометацин не оказывал существенного влияния и на концентрацию фибриногена плазмы.
Показатели тромбоэластограмм у животных также свидетельствовали о нормализации большинства ее параметров. Вместе с тем показатель Т не достигал значений исходного уровня. Этот факт наряду с повышением уровней РКФМ свидетельствует о том, что к исходу суток у животных еще остаются проявления гипокоагуляции. Наш материал не позволяет уточнить причину этой гипокоагуляции. Вероятно, за проявления гипокоагуляции ответственны процессы фибринолиза, в том числе и неферментного. Возможно, неферментный фибринолиз не зависит от функциональной активности простагландинов, так как торможение синтеза последних не влияло на уровень гипокоагуляции.
Проведенные исследования убедительно свидетельствуют о том, что введение индометацина животным с экспериментальной эндотоксинемией сопровождается предотвращением развития либо быстрым купированием ее проявлений (клинических и со стороны гемостаза).
При исследовании коагулограммы и тромбоэластограммы у животных с местной реакцией Шварцмана, которым предварительно вводили индометацин, отмечена совершенно иная динамика показателей гемостаза. В этой группе животных не выявлена фаза гиперкоагуляции после инъекции 1-й дозы эндотоксина или при местной реакции Шварцмана. Большинство параметров коагулограммы и тромбоэластограммы отражали умеренную тенденцию к гипокоагуляции или не отличались от исходных значений.
Инъекция 2-й дозы эндотоксина при местной реакции Шварцмана не вызывала гипокоагуляцию. В ряде случаев, наоборот, показатели Коагулограммы (тромбиновое время) достоверно свидетельствовали о повышении свертывающего потенциала крови. Большинство показателей тромбоэластограммы не отличались от исходных значений. Обращало на себя внимание и то, что в условиях подавления синтеза простагландинов на месте внутрикожного введения эндотоксина некротические процессы не развивались. Таким образом, индометацин блокировал развитие местной реакции Шварцмана.
При воспроизведении генерализованной реакции Шварцмана на фоне индометацина установлены аналогичные изменения в состоянии гемостаза. Так, на введение 1-й дозы эндотоксина в этой группе животных не выявлена фаза гиперкоагуляции. Показатели коагулограммы и тромбоэластограммы были характерными для состояния умеренной гипокоагуляции или не отличались от исходных значений.
Инъекция 2-й дозы эндотоксина на фоне однократного введения индометацииа не сопровождалась гипокоагуляцией. Большинство параметров гемостаза не отличалось от исходных значений, кроме показателя концентрации фибриногена, который повышался (Р<0,05). При введении индометацииа все экспериментальные животные выживали.
Следует подчеркнуть, что уровни фибриногена при местной и генерализованной реакциях Шварцмана достоверно выше значений этого показателя при однократной инъекции эндотоксина.
Причины этого явления остаются неясными. Вероятно, при воспроизведении местной и генерализованной реакций Шварцмана определенное значение имеют факторы гиперчувствительности замедленного типа, что и отражается на этом показателе.
Результаты исследований дают основание считать, что развитие местной и генерализованной реакций Шварцмана тесно связано с резким усилением синтеза простагландинов.
Таким образом, результаты наших исследований состояния гемостаза при однократном и двукратном введениях эндотоксина позволяют сделать вывод о том, что свойство эндотоксина вызывать нарушения в системе гемостаза определяется его способностью резко усиливать синтез простагландинов. Нами не выявлено принципиальной разницы в изменении состояния гемостаза при одно- и двукратном введениях эндотоксина. Назначение животным индометацииа, вызывающего угнетение синтеза простагландинов, сопровождалось совершенно иной динамикой показателей гемостаза при всех вариантах введения эндотоксина.
В этой связи важное значение приобретало изучение состояния микроциркуляции различных органов, а также эндокринного аппарата почек на фоне введения индометацииа. Введение индометацииа интактным животным не приводило к каким-либо изменениям в почках, печени и тонкой кишке, за исключением снижения плотности гранул в эпителиоидных клетках юкстагломерулярного аппарата при электронно-микроскопическом его исследовании. В интерстициальных клетках мозгового вещества почек через 3 ч после введения индометацииа отмечено накопление, а через 24 ч – уменьшение количества липндных гранул. Однако эти изменения не были статистически достоверными.
Введение индометацина через 1 ч после внутривенного введения эндотоксина сопровождалось быстрым купированием клинических проявлений сальмонеллезной эндотоксинемии. У животных прекращался жидкий стул, нормализовалась температура. При микроскопии через 24 после введения эндотоксина были обнаружены умеренно выраженные изменения почек, напоминающие таковые, выявленные через 3 ч после введения эндотоксина (рис. 9). Количество гранул в интерстициальных клетках и плотность гранул в эпителиоидных клетках юкстагломерулярного аппарата мало отличались от нормы.
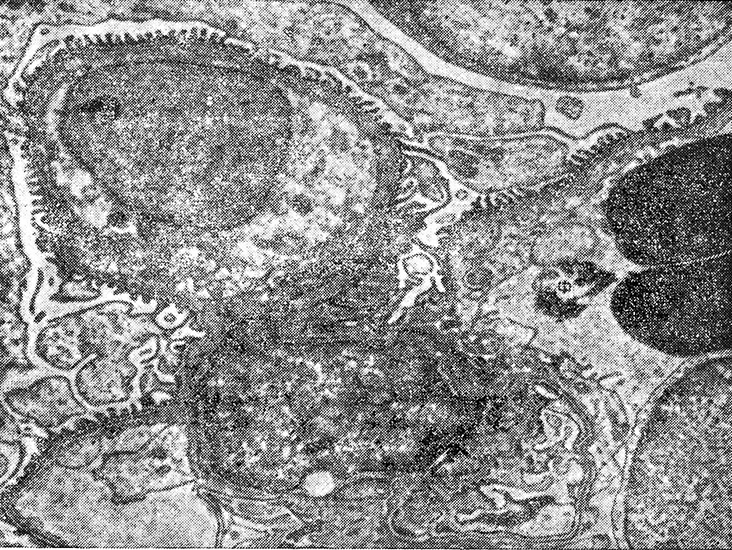
Рис. 9. Введение индометацина через 1 ч после внутривенного введения сальмонеллезного эндотоксина.
Незначительные скопления фибрина (Ф) в просвете капилляра почечного клубочка. х 6000.
Предварительное введение индометацина за 1 ч до внутривенного введения эндотоксина предотвращало развитие сальмонеллезной интоксикации. У животных не развивалась диарея, не повышалась температура. Степень морфологических изменений не достигала уровня, выявленного в аналогичные сроки у животных, которым индометацин не назначался.
Проведенные исследования свидетельствуют о том, что сальмонеллезная эндотоксинемии существенным образом влияет на эндокринный аппарат почек, повреждает микроциркуляцию и структуру исследованных органов. Наши данные совпадают с результатами исследований О. Р. Кузнецовой (1976), McKay D. G. и соавт. (1966), показавших, что сальмонеллезная интоксикация приводит к дистрофическим изменениям, интерстициальному отеку и клеточной инфильтрации во многих органах, включая почки, тонкую и толстую кишку.
В результате наших исследований не получены достоверные данные о вовлечении интерстициальных клеток мозгового вещества почек, которые являются одним из источников простагландинов в органе, в реализации биологического действия эндотоксина сальмонелл. По нашему мнению, это связано с особенностями распределения эндотоксина в организме животных. При специальном изучении этого вопроса было установлено, что в первые часы после внутривенной инъекции эндотоксин обнаруживается в печени, затем в легких, но в почках он не был обнаружен [Freudenberg М. et al., 1980]. Видимо, простагландины, синтезируемые в интерстициальных клетках мозгового вещества почек, реализуются местно, участвуя в регуляции сосудистого русла почек.
При изучении морфологических изменений в почках животных при местной и генерализованной реакциях Шварцмана установлено, что введение индометацииа предотвращало появление фибриновых тромбов в капиллярах почек. Степень выраженности других морфологических изменений на фоне индометацина была более умеренной.
Следует отметить, что морфологические измененпя, возникающие при эндотокеинемии, обусловлены как прямым действием эндотоксина, так и нарушениями микроциркуляции. Введение индометацина способствует как ослаблению эндотокеинемии, так и нормализации микроциркуляции.
Определенный интерес представляют результаты экспериментальных исследований группы ученых, изучавших влияние повышенных уровней простагландинов на состояние структуры тканей. Так, по данным Л. А. Энтсик (1982), инъекция простагландинов крысам приводит к усилению тромбообразования, а назначение индометацина снижает его. В работе С. И. Пушкарь и соавт. (1982) показано, что под влиянием простагландина Ег в культуре клеток печени происходили отчетливые изменения ультраструктуры гепатоцитов. Характер этих изменений чрезвычайно близок к описанным нами при эндотоксинемии. Индометацин, по данным С. И. Пушкарь и соавт. (1982), обладает способностью предотвращать развитие этих ультраструктурных изменений.
Таким образом, анализ результатов и многочисленных исследований дает основание полагать, что развитие структурных нарушений и тромбогеморрагических осложнений при сальмонеллезе связано с резким усилением синтеза простагландинов.
В связи с очевидной ролью эндотоксина сальмонелл в развитии стереотипных изменений гемостаза у животных есть основание предположить, что они являются составной частью синдрома сальмонеллезной интоксикации, включающего комплекс клинических и патофизиологических проявлений, сопровождающих экспериментальную эндотоксинемию.
Чрезвычайная близость экспериментальной сальмонеллезной интоксикации к клиническим проявлениям гастроинтерстициальной формы сальмонеллеза позволяет в известной степени экстраполировать результаты экспериментальных исследований на клинические наблюдения.
В эксперименте нами показано, что патогенез плейотропного действия эндотоксина связан с его способностью стимулировать биосинтез простагландинов из арахидоновой кислоты в условиях in vitro и in vivo. Эти факты дают основание считать, что патогенез интоксикации при сальмонеллезе связан с ответной реакцией макроорганизма, заключающейся в резком усилении под влиянием эндотоксина синтеза мощных тканевых биорегуляторов – простагландинов.
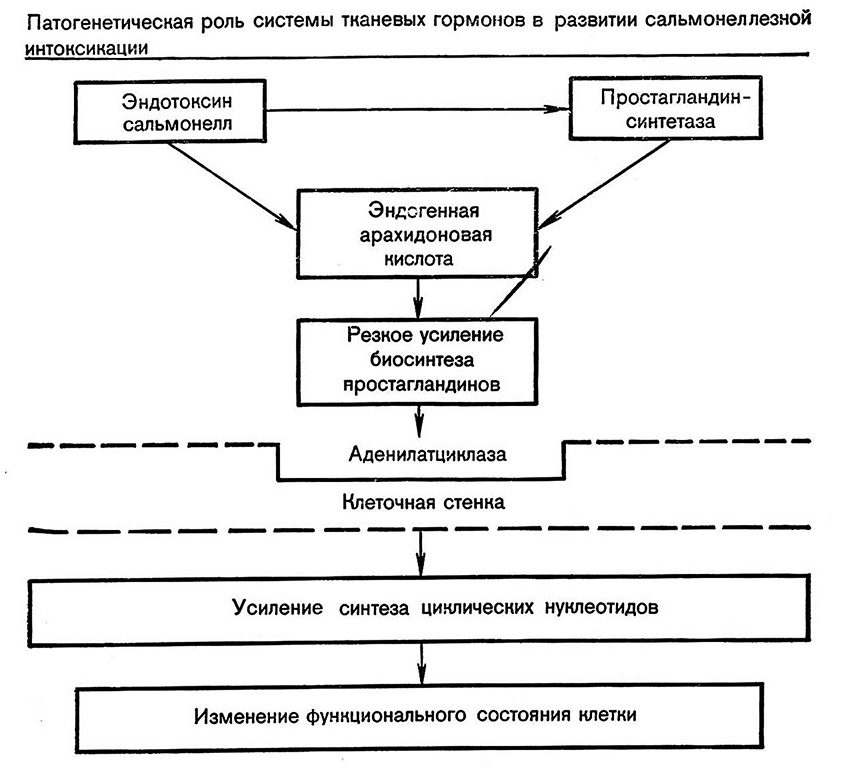
Нами предложена концепция о патогенетической роли простагландинов в развитии клинических проявлений сальмонеллеза и нарушений гемостаза, микроциркуляции и деятельности сердечно-сосудистой системы. С учетом известных данных о роли нервной, гормональной и других систем организма, осуществляющих адаптационно-приспособительные реакции макроорганизма, роль тканевых гормонов в развитии сальмонеллезной интоксикации представлена на схеме 2.
Схема 2
Учитывая результаты собственных экспериментальных исследований и данные о широком применении индометацина в клинике, мы предлагаем включить индометацин в схему комплексного лечения больных гастроинтестинальной формой сальмонеллеза.
