В конце 70-х годов в биохимии был разработан ряд новых методологических подходов, ознаменовавших начало эры генетической инженерии. Методы рекомбинантных ДНК, перевернувшие наши представления о возможностях биотехнологии в сфере удовлетворения потребностей человека, позволяют направленно конструировать генетический материал, вводить его в живые клетки и с помощью последних реализовать эту генетическую информацию. Кроме того, благодаря технологии рекомбинантных ДНК значительно ускорились темпы расширения и развития наших знаний о функциях ДНК, организации генов, регуляции их экспрессии, первичной структуре белков, что в свою очередь создает основу для понимания природы различных заболеваний и борьбы с ними. Возможность введения любых сегментов ДНК в клетки позволяет создавать промышленные микроорганизмы, способные синтезировать ценнейшие белки. В этом разделе мы уделим основное внимание применению генетической инженерии в промышленности и использованию в качестве организма-хозяина бактерии Escherichia coli.
Прежде чем перейти к изучению основных приемов и методов генетической инженерии, целесообразно рассмотреть общие принципы процесса и познакомиться со специальной терминологией. Ha рис. 6.21 изображена упрощенная схема клонирования фрагмента ДНК. Под клонированием здесь мы понимаем приготовление колонии генетически идентичных клеток, которые содержат интересующий нас сегмент ДНК. Этот сегмент, названный на рис. 6.21 «чужеродной ДНК», получают путем разрезания большого участка ДНК на фрагменты с помощью специфичных эндонуклеазных ферментов или путем синтеза – химического или ферментативного. Чужеродную ДНК соединяют Un vitro) с вектором – специальным инструментом для введения чужеродной ДНК в бактериальную клетку. B нашем примере вектор представляет собой бактериальную плазмиду, неоколько модифицированную с целью облегчения процесса клонирования. Затем молекулу рекомбинантной ДНК (комплекса чужеродной ДНК с плазмидой) вводят в ДНК клетки бактерии, используя механизм трансформации. После этого с помощью описанных ниже методов идентифицируют клон, содержащий плазмиду с чужеродной ДНК.
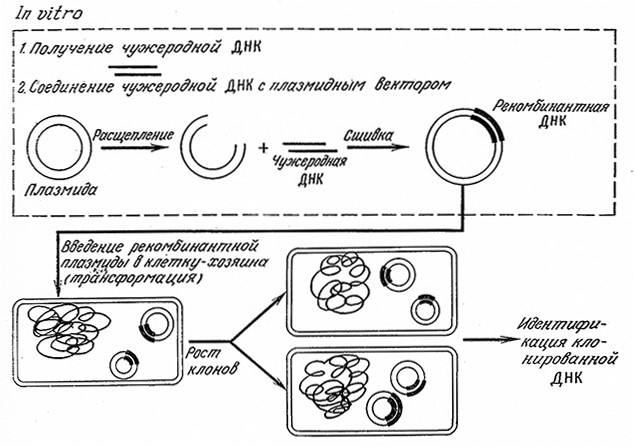
РИС. 6.21. Основные этапы клонирования фрагментов чужеродной ДНК.
Клонирование фрагмента ДНК дает возможность получить его в количествах, достаточных для детального изучения и применения в качестве реагента в последующих биохимических и генетических работах. B качестве примера использования клонированных ДНК для аналитических целей можно указать на ход работ по изучению интерферона из фибробластов (IFN-β) в конце 70-х годов. Этот белок был чрезвычайно труднодоступен, поэтому даже с помощью самых чувствительных приборов для определения аминокислотной последовательности удавалось выяснить природу только нескольких N-концевых аминокислотных остатков белка. Однако вскоре после клонирования гена интерферона из фибробластов была определена его нуклеотидная последовательность, из которой с учетом генетического кода непосредственно вытекала и аминокислотная последовательность соответствующего белка. B разд. 6.4.4 мы рассмотрим примеры использования клонированных ДНК для получения штаммов микроорганизмов, продуцирующих большие количества чужеродных белков, а следующий раздел будет посвящен изучению ряда ферментов, без которых невозможна технология рекомбинантных ДНК.
6.4.1. Ферменты для расщепления и сшивки ДНК
Успехи генетической инженерии во многом связаны с выделением и последующим промышленным производством ряда ферментов, с помощью которых можно разрезать, видоизменять и соединять молекулы ДНК в лабораторных условиях. Особое место среди этих ферментов занимают рестрикционные эндонуклеазы, способные узнавать и расщеплять специфические нуклеотидные последовательности в молекулах ДНК. Например, реетриктаза EcoiRI специфична по отношению к последовательности шести нуклеотидных остатков в двойной спирали следующего типа:
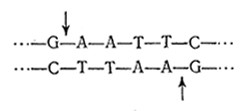
Эта рестриктаза расщепляет каждую цепь между остатками A и G по связям, отмеченным стрелками. B результате образуются два фрагмента:
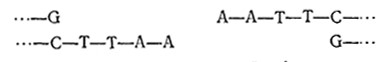
Обратите внимание на то, что каждый фрагмент содержит короткую одноцепочечную последовательность, называемую «липким концом», поскольку эти две последовательности комплементарны и, следовательно, имеют тенденцию к связыванию друг с другом за Счет водородных связей между парными основаниями. Реализация этой тенденции зависит от экспериментатора, который посредством изменения рН или температуры может добиться образования водородных связей или их разрушения.
Таблица 6.7. Некоторые рестрикционные эндонуклеазы, соответствующие сайты узнавания H расщепляемые связи (указаны стрелками)
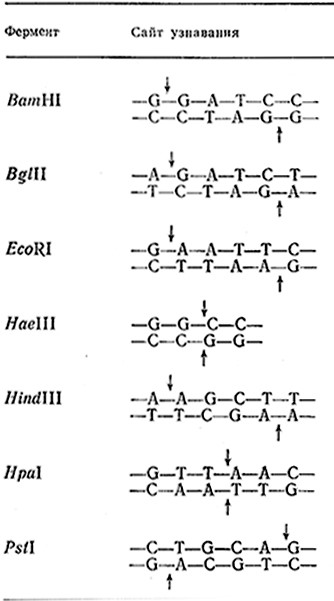
К настоящему времени идентифицированы более 100 различных ферментов, обладающих рестриктазной активностью. В табл. 6.7 перечислены наиболее часто используемые рестриктазы, соответствующие сайты узнавания и гидролизуемые ими фосфодиэфирные связи. Нетрудно заметить, что не все рестриктазы образуют липкие концы. Рестриктаза Hpal, например, расщепляет специфическую последовательность шести нуклеотидных остатков таким образом, что образуются только двухцепочечные фрагменты с «тупыми концами».
Номенклатура рестрикционных ферментов базируется на названии организма, из которого данная рестриктаза была впервые выделена. Первая прописная буква в обозначении рестрик- тазы соответствует первой букве названия рода организма, а последующие две или три строчные буквы берутся из первых двух или трех букв названия вида этого организма. Наконец, римская цифра обозначает порядковый номер фермента, отражающий хронологию его выделения из данного организма. Так, рестриктаза Bglll представляет собой вторую рестриктазу, обнаруженную в бактерии Bacillus globigii. Для уточнения природы штамма, продуцирующего данный фермент, в название рестриктазы иногда включают дополнительные буквы; так, рестриктаза ZfcoRI была выделена из E. coli RY13.
Ценность рестрикционных ферментов в технологии рекомби- нантных ДНК обусловливается их высокой специфичностью, т. е. способностью расщеплять только строго определенные нуклеотидные последовательности. Поскольку вероятность наличия в молекуле ДНК специфических сайтов узнавания, содержащих от 4 до 6 нуклеотидных остатков, не очень велика, то образующиеся в результате гидролиза рестриктазами фрагменты ДНК обычно имеют довольно большую молекулярную массу (как правило, несколько сотен нуклеотидных остатков). Это достаточно много для того, чтобы образовавшиеся фрагменты несли ценную генетическую информацию, и одновременно довольно мало для того, чтобы их можно было изучать и модифицировать in virto.
Объем настоящего учебною пособия позволяет лишь вкратце упомянуть другие важные ферменты, применяющиеся в технологии рекомбинантных ДНК. В первую очередь следует отметить ДНК-лигазу. Предположим, что мы создали такие условия, когда липкие концы, образовавшиеся в результате расщепления различных молекул ДНК под действием ZfcoRI5 «отжигаются», т. е. сближаются и связываются за счет образования пар оснований. Образовавшиеся ДНК не будут, однако, кова- лентно замкнутыми, поскольку между остатками AhGb обеих цепях отсутствуют фосфодиэфирные связи. Последние могут быть вновь созданы с помощью ДНК-лигазы, катализирующей конденсацию З'-гидроксильной группы с 5'-фосфатной группировкой.
Таким путем, например, отожженные фрагменты
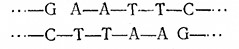
ковалентно сшиваются ДНК-лигазой:
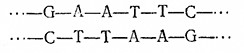
В заключение отметим другие ферменты, катализирующие синтез олигонуклеотидов (рис. 6.22). На матричной цепи ДНК фермент ДНК-полимераза I катализирует построение комплементарной цепи в 5' → З'-направлении. Для осуществления этого процесса необходимо присутствие всех четырех дезоксприбонуклеотидов, а также праймера, обладающего свободной З'-гидроксильиой группой. Обратная транскриптаза (ревертаза; РНК-зависимая ДНК-полимераза) выполняет задачу, потребность в решении которой возникает сравнительно редко; этот фермент как бы обращает обычный порядок передачи генетической информации, синтезируя цепь ДНК, комплементарную матрице мРНК. Соединение различных фрагментов ДНК часто облегчается после их удлинения на комплементарные гомополи- мерные цепи (хвосты); в присутствии только одного нуклеотид- трифосфата фермент концевая трансфераза способен много-кратно присоединять этот нуклеотид к концевой З'-ОН-группе .молекулы ДНК.
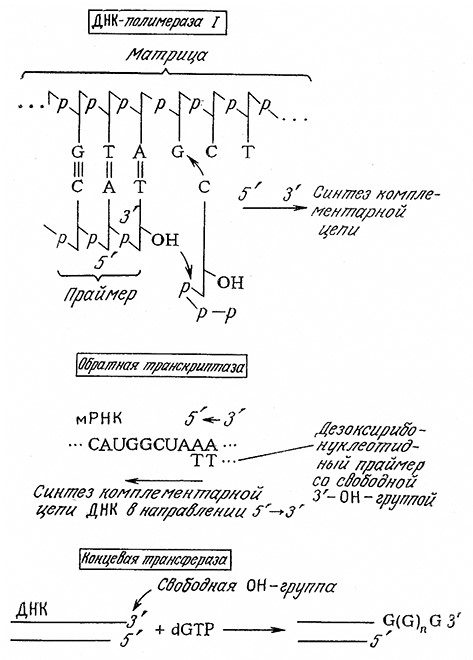
РИС. 6.22. Характерные реакции некоторых синтезирующих ферментов, используемых
при получении рекомбинантных ДНК.
Некоторые другие ферменты, играющие большую роль в методологии генетической инженерии, мы упомянем ниже, а следующий раздел будет посвящен требованиям, предъявляемым к векторам рекомбинантных ДНК.
6.4.2. Векторы для Escherichia coli
Векторами называют молекулы ДНК, обеспечивающие амплификацию фрагмента ДНК в растущей популяции клеток. Чтобы вектор отвечал всем предъявляемым к нему в процессе клонирования требованиям, он должен обладать следующими свойствами:
1. Способностью реплицироваться в клетке-хозяине.
2. Способностью включать чужеродные ДНК различной молекулярной массы без нарушения способности к репликации.
3. Легкостью введения в клетку-хозяина после включения чужеродной ДНК.
4. Наличием генетического маркера селекции, обеспечивающего быструю положительную селекцию клеток, содержащих вектор.
5. Наличием только одного сайта, подвергающегося расщеплению одной или несколькими ресгрикционными эндонуклеа- зами.
Для клонирования ДНК в Е. coli применяли два класса векторов– плазмиды и бактериофаги. Здесь основное внимание будет уделено плазмидным векторам.
Как мы уже упоминали выше, при введении плазмид в клетки Е. coli используется механизм трансформации. Поскольку трансформированная клетка Е. coli может включить только одну плазмиду, а для последующего выделения, идентификации и применения интересующих нас последовательностей ДНК необходим клон, состоящий из большого числа идентичных клеток, содержащих плазмиды, то плазмида обязательно должна обладать способностью к репликации в растущей бактериальной клетке. Для этого в свою очередь нужно, чтобы в состав плазмиды входил участок начала репликации – нуклеотидная последовательность (около 600 пар оснований в случае плазмид Е. coli типа ColE-I), направляющая и регулирующая процесс репликации таким образом, чтобы каждая клетка содержала достаточное число копий плазмид (обычно около 30).
Немаловажны и генетические маркеры селекции. Если, например, в плазмиду включить ген устойчивости к антибиотику, то быстрая положительная селекция содержащих плазмиды клеток lie вызовет затруднений. Для этого достаточно вырастить клетки на среде, содержащей антибиотик в такой концентрации, которая приведет к гибели всех не имеющих плазмиды клеток, но в то же время обеспечит рост клеток, содержащих плазмиды, а следовательно, и ген устойчивости к антибиотику. Другой общий подход к селекции заключается в использовании мутантной клетки-хозяина, у которой отсутствует фермент, необходимый для роста в определенной среде, и в одновременном введении соответствующего этому ферменту нормального гена в рекомбинантную плазмиду. В этом случае экспрессия гена плазмиды комплементирует генетический дефект клетки-хозяина.
В технологии рекомбинантных ДНК маркеры селекции в плазмидах могут выполнять две различные функции. Во-первых, методы положительной селекции, позволяющие быстро идентифицировать содержащие вектор колонии, резко интенсифицируют лабораторные исследования. Во-вторых, при последующем выращивании содержащих плазмиду клеток давление отбора (обусловленное, например, введением в питательную среду антибиотика) сводит к минимуму конкуренцию со стороны любых не содержащих плазмиду клеток, которые могут возникать входе роста популяции.
Плазмидные векторы для клонирования в Е. coli особенно широко изучались в последние годы. К числу наиболее популярных относится плазмида pBR322 (рис. 6.23), содержащая ряд единичных сайтов рестрикции, гены устойчивости к тетрациклину и ампициллину, а также участок начала репликации, обеспечивающий амплификацию плазмиды в клетке. Здесь под амплификацией (экстракопированием) понимается существен-ное повышение количества плазмид по отношению к хромосомной ДНК. В случае pBR322 и родственных плазмид амплификация становится возможной благодаря тому обстоятельству, что в отличие от хромосомы репликация плазмиды в клетке-хозяине может осуществляться и тогда, когда белковый синтез остановлен. Поэтому добавление к культуральной жидкости ингибитора белкового синтеза, например хлорамфеникола, приводит к синтезу от 30 до 1000 и более копий плазмиды, а следовательно, и клонируемой ДНК. Это позволяет получить последнюю в количествах, достаточных для ее идентификации и последующего использования в качестве реагента при конструировании других ДНК. В плазмиде pBR322 и родственных векторах могут клонироваться сегменты чужеродных ДНК, содержащие до 15 тысяч пар оснований.
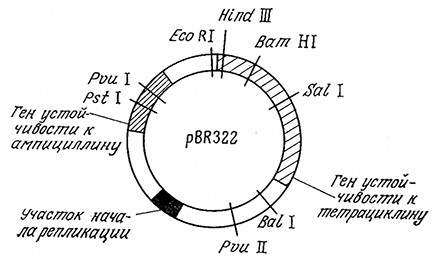
РИС. 6.23. Генетическая карта плазмиды pBR322. Показаны локусы генов и ряд сайтов рестрикции.
Расшифрована последовательность всех 4363 пар оснований плазмиды.
В качестве клонирующего вектора широко изучался также бактериофаг λ, который сохраняет способность к репликации и литические функции после делеции 25% генома дикого типа; это позволяет клонировать сегменты чужеродной ДНК, содержащие до 12 тысяч пар оснований. Значительно большие сегменты ДНК (до 50 тысяч пар оснований) удается клонировать с помощью гибридных плазмидно-фаговых λ векторов, называемых космидами; последние могут быть введены в головку и хвост фага λ. По этой причине векторы-бактериофаги предпочтительнее в тех случаях, когда возникает необходимость в клонировании библиотек, состоящих из целого генома эукариоты или из ее очень больших фрагментов. Полезные векторы получали также из фагов с одноцепочечным геномом, например из фага M13. Получаемые с их помощью одноцепочечные клоны особенно удобны для определения нуклеотидных последовательностей ДНК по методу Сэнджера. Следующий раздел будет посвящен методам выяснения последовательностей нуклеотидных остатков идругим методам идентификации клонированных ДНК.
6.4.3. Идентификация клонированных ДНК
Предположим, что рекомбинантные плазмиды удалось включить в популяцию клеток Е. coli и что содержащие плазмиды клетки удалось обнаружить на агаровых пластинках в селективной среде. В этом разделе мы вкратце рассмотрим методы скрининга полученных таким путем клонов и детального изучения инсерций (вставок) ДНК из интересующих нас клонов.
Прежде всего нам нужно отличать клоны, содержащие плазмиды с инсерциями чужеродной ДНК, от клонов, в состав которых входят только исходные плазмиды; это может быть достигнуто с помощью инсерционной инактивации в процессе клонирования, Предположим, например, что фрагменты ДНК включены в сайт плазмиды pBR322, расщепляемый рестриктазой PstL Поскольку инсердия в этом центре разрывает ген устойчивости к ампициллину, то содержащие рекомбинантные плазмиды клетки будут чувствительны к ампициллину и устойчивы к тетрациклину. Напротив, клетки с плазмидами без инсерции будут устойчивы как к ампициллину, так и к тетрациклину.
В этом случае клоны, содержащие рекомбинантные плазмиды, можно обнаружить следующим образом. Сначала клетки выращивают на пластинках в присутствии тетрациклина; таким путем избавляются от существенного фона не содержащих плазмиды «леток. Затем применяют метод реплик (отпечатков) войлочную подушечку или нитроцеллюлозную фильтровальную бумагу прижимают сначала к исходной матричной пластинке, а затем ко второй пластинке (реплике), получая на ней точную копию распределения колоний на матричной пластинке. Если реплика обработана ампициллином, то колонии клеток, содержащих плазмиды с инсерциями в гене устойчивости к ампициллину, растут на матричной пластинке, но не растут после перепечатки.
Поиск в клоне специфических нуклеотидных последовательностей может быть осуществлен с помощью ряда методик, основанных на гибридизации денатурированной (т. е. одноцепочечной) ДНК из клона с особым нуклеотидным зондом, обычно меченным радиоактивным изотопом 32P. Нуклеотидная последовательность зонда (которым может быть как ДНК, так и мРНК) комплементарна части последовательности искомого сегмента ДНК-Зонды мРНК получают путем выделения мРНК из клеток, обогащенных интересующей исследователя генетической информацией, а зонды ДНК выделяют на одной из первых стадий «лонирования или получают прямым химическим синтезом.
В методе гибридизации колонии in situ на нитроцеллюлоз ной сЬильтровальной бумаге, лежащей на чашке Петри с питательной средой, выращивают реплику клонов. Затем клетки разрушают лизоцимом, а ДНК денатурируют обработкой NaOH, после чего нитроцеллюлозную бумагу отделяют, обрабатывают зондом, промывают и экспонируют с рентгеновской пленкой. На пленке появляются темные пятна в тех местах, где произошла гибридизация зонда с комплементарными последовательностями ДНК; эти клоны и представляют интерес для дальнейшего изучения.
Плазмидную ДНК, которую можно выделить ультрацентрифугированием в градиенте плотности хлорида цезия, далее обрабатывают рестриктазой. Полученные фрагменты ДНК образуют своеобразную картину («отпечатки пальцев») для данной илазмидной ДНК. Фрагменты могут быть разделены в соответствии с их молекулярными массами методом электрофореза в агарозном геле (0,5–1,5%); для обнаружения фрагментов ДНК часто используют флуоресцентный (краситель бромистый этидий, специфичный по отношению к двухцепочечным нуклеиновым кислотам. Путем сравнительного анализа длины фрагментов, образующихся при действии других рестриктаз и при совместном расщеплении несколькими рестрикционными эндонуклеазами, получают рестрикционную карту, характеризующую относительные положения различных сайтов рестрикции. Подобные карты чрезвычайно полезны для проверки правильности включения ДНК в заданные участки плазмиды и в качестве основы для дальнейшей работы с рекомбинантными плазмидами.
Упоминавшиеся выше меченные радиоактивными изотопами зонды можно использовать и для идентификации определенных фрагментов, образующихся при обработке рестриктазами. В методе блоттинга по Саузерну фрагменты ДНК в геле денатурируют щелочью и на гель накладывают нитроцеллюлозную плен'ку. Сверху помещают фильтровальную бумагу, в которую переходит буферный раствор; одновременно фрагменты ДНК переходят на нитроцеллюлозную пленку. ДНК ковалентно связывается с нитроцеллюлозой при 80°С. Затем связанные денатурированные фрагменты ДНК обрабатывают, как было описано выше, зондом, экспонируют с рентгеновской пленкой и таким образом идентифицируют полосы на электрофореграмме, соответствующие комплементарным зонду нуклеотидным последовательностям.
Современные экспериментальные методы биохимии ДНК настолько чувствительны, что нескольких нанограммов ДНК, выделенных из полосы на гелевой электрофореграмме, достаточно для клонирования или для дальнейшего изучения. Очевидно, что исчерпывающая идентификация фрагмента ДНК должна включать в себя определение его нуклеотидной последовательности. В 70-е годы были разработаны два метода быстрого, надежного и эффективного определения последовательностей нуклеотидных оснований в ДНК – Максама – Гилберта и Сэнджера. Эти методы оказали громадное воздействие на биологические науки вообще и на биотехнологию в частности.
В методе Максама – Гилберта двойную спираль ДНК сначала метят, присоединяя радиоактивную метку к одному концу каждой цепи. Затем ДНК денатурируют и препарат одной из двух цепей разделяют на четыре аликвотные части, каждую из которых обрабатывают специфичным реагентом. В результате каждой из таких обработок селективно разрушается одно основание (или два), а цепи ДНК в этих центрах расщепляются. Для успеха метода существенно, чтобы это разрушение было·частичным; в идеальном варианте желательно, чтобы каждая цепь расщеплялась бы только в одном центре. Затем образовавшиеся в результате таких четырех параллельных расщеплений фрагменты изучают на достаточно протяженном полиакриламидном геле. Полосы в геле обнаруживают с помощью чувствительной к рентгеновскому излучению фотопленки. Определяемую нуклеотидную последовательность считывают непосредственно с четырех параллельных дорожек гелевой электрофореграммы (рис. 6.24). Таким методом в одном эксперименте без труда можно выяснить последовательность около 200 нуклеотидных остатков. Метод определения нуклеотидных последовательностей по Сэнджеру рассмотрен в работе [21]; обычно его применяют при изучении более крупных фрагментов ДНК.
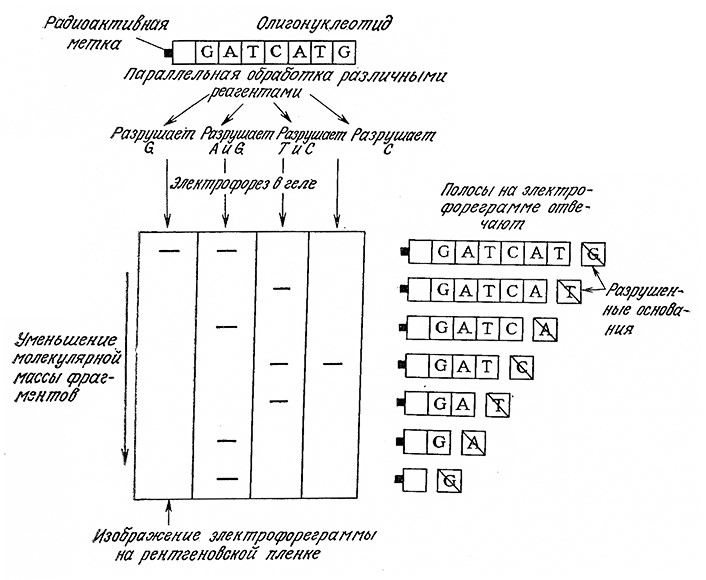
РИС. 6.24. Определение нуклеотидной последовательности методом Максама – Гилберта. При обработке изучаемой последовательности
четырьмя специфическими реагентами, разрушающими различные основания, образуется смесь фрагментов, молекулярные массы которых
зависят от положения расщепленного основания. Расположение полос на четырех дорожках гелевой электрофореграммы позволяет
считывать нуклеотидную последовательность непосредственно с геля; считывание сверху вниз соответствует направлению
последовательности к меченому концу исходного олигонуклеотида.
Часто возникает необходимость в скрининге из множества различных клонов одного клона (или нескольких клонов), обладающих способностью к экспрессии определенного белка. В большинстве случаев для решения такой задачи прибегают к помощи антитела, специфичного по отношению к данному белку. В антитела вводят радиоактивную метку (например, 125I) и раствором антител обрабатывают изучаемые колонии на пластинке; затем их промывают и с помощью рентгеновской пленки обнаруживают искомые колонии, содержащие данный белок. В другом варианте антитела связывают с ферментом и антигены обнаруживают по свойствам этого фермента. Идентифицировать колонии, содержащие искомый белок, можно, например, с помощью хромо-генного субстрата (т. е. субстрата, изменяющего свою окраску или окрашивающегося после обработки ферментом). Более чувствительные иммунохимические методы, с помощью которых можно обнаружить белки в концентрациях 1–5 молекул в клетке Е. coli, описаны в приведенной в конце: главы литературе.
Объем и тематика этой книги не позволяют обсудить детально все методы идентификации и изучения клонов; все же надо надеяться, что даже приведенные здесь данные дают* представление о том, насколько трудоемкой, длительной и утомительной может быть работа по скринингу клонов и по контролю каждого шага в построении новых рекомбинантных молекул. Хотя для специалиста в области биохимической технологии больший интерес представит тема следующего раздела, посвященного экспрессии клонированных генов, следует отдавать себе отчет в том, что основным препятствием в программе синтеза чужеродного белка в Е. coli или другой клетке-хозяине скорее всего будет именно клонирование гена. В то же время успехи методических разработок и развитие их автоматизации; будут содействовать внедрению «типовых методик» молекулярной генетики, что в свою очередь создаст основы для существенного расширения набора доступных и интересных генов и регуляторных последовательностей.
6.4.4. Экспрессия эукариотических белков в Е. coli
Первыми примерами успешного применения генетической инженерии в биотехнологии были синтезы ряда ценных белков· эукариот в клетках Е. coli. В этом разделе мы прежде всего рассмотрим методы получения генов, пригодных для экспрессии эукариотических белков, затем обсудим требования, предъявляемые к вектору экспрессии, и завершим раздел рядом замечаний, касающихся проблем скрининга продуктов экспрессии генов, а также устойчивости генов и соответствующих белков.
Если вернуться к рис. 6.7 и соответствующему разделу текста, нетрудно понять, что прямая экспрессия генов эукариот Е. coli связана с одним принципиальным препятствием. Прокариоты вообще и Е. coli в частности не обладают биохимическим механизмом, способным осуществлять сплайсинг РНК. Поэтому эукариотический ген, включенный непосредственно в Е. coli (при условии рассматриваемой ниже соответствующей бактериальной регуляции экспрессии), не будет нормально экспрессироваться. В Е. coli будут транслироваться последовательности интронов эукариотического гена мРНК и синтезирующийся полипептид в общем случае будет резко отличаться от нормального эукариотического белка.
Очевидно, нам нужно каким-то образом приготовить бактериальный ген, отвечающий эукариотическому белку, т. е. такой ген, который содержал бы только нуклеотидную последовательность, кодирующую первичную структуру белка, и не имел бы ни одного интрона. Эта цель может быть достигнута двумя принципиально различными путями. Один из них сводится к химическому синтезу заданной нуклеотидной последовательности. Хотя здесь мы не можем углубляться в детали химического синтеза генов (интересующийся этой проблемой читатель может найти все необходимые сведения в литературе), все же необходимо подчеркнуть, что синтетическая химия ДНК пока еще не позволяет осуществлять прямой синтез всего гена. Обычно синтезируют олигонуклеотиды с частично перекрывающимися последовательностями, которые обладают способностью к самосборке в полную двухцепочечную нуклеотидную последовательность за счет образования водородных связей в парах оснований. В качестве примера на рис. 6.25 приведен ген, кодирующий последовательность 21 аминокислотного остатка цепи А инсулина человека; этот ген был собран из 12 различных олигодезоксирибонуклеотидов, построенных из 10–15 нуклеотидных остатков. При планировании синтеза гена следует принимать во внимание и ряд других требований, связанных с клонированием и экспрессией, в том числе свойства кодонов, отсутствие сигналов терминации, наличие сайтов рестрикции.
Возможность химического синтеза генов или их фрагментов создает базу для чрезвычайно перспективной области белковой инженерии. В принципе уже сейчас мы можем заменить любую аминокислоту в белке, с тем чтобы попытаться повысить биологическую активность или технологическую устойчивость данного белка. Более того, мы можем заставить клетку синтезировать новые (или, во всяком случае, неизвестные нам) аминокислотные последовательности и таким путем создавать новые катализаторы, лекарственные препараты и компоненты пищевых продуктов. К сожалению, в настоящее время мы знаем так мало о связи между первичной структурой и функцией белков, что возможности улучшения свойств белков путем их модификации пока что остаются практически нереализованными.
В другом пути получения бактериального гена, соответствующего эукариотическому белку, исходят из зрелой эукариотической матричной РНК, уже прошедшей стадию сплайсинга.
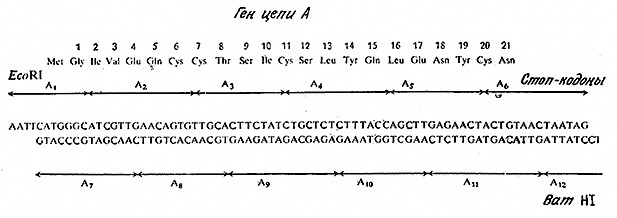
РИС. 6.25. Ген, отвечающий цепи А инсулина человека (аминокислотная последовательность приведена в верхней части рисунка).
Этот ген был синтезирован нз 12 олигодезоксирибонуклеотидов A1–А12.
Как схематично показано на рис. 6.26, соответствующая необходимому белку мРНК сама может выполнять роль матрицы, на которой особый фермент, обратная транскриптаза, может синтезировать комплементарную цепь ДНК. Цепи мРНК и ДНК затем разделяют, на цепи ДНК с помощью ДНК-полимеразы строят комплементарную цепь, после чего специфическим ферментом нуклеазой S1 отщепляют небольшой петлевой одноцепочечный участок. В результате образуется молекула двухцепочечной комплементарной ДНК (кДНК). К последней, как показано на рис. 6.26, с помощью концевой трансферазы присоединяют гомополимериые последовательности дезоксицитидина (dC). Таким путем создаются предпосылки для последующего клонирования, предпочтительно с плазмидным вектором pBR322, расщепленным рестриктазой PstI и имеющим гомополимерные последовательности (dG).
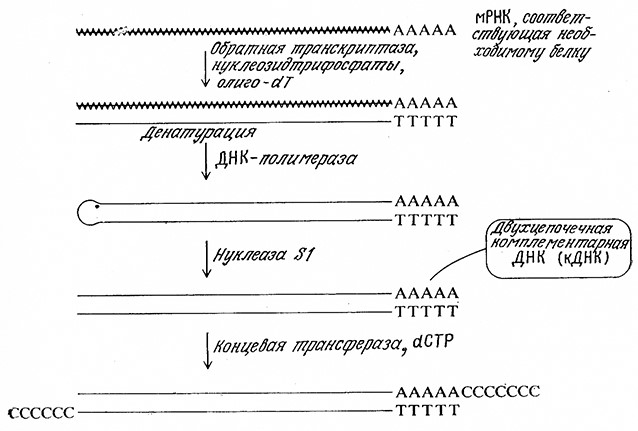
РИС. 6.26. Схема синтеза двухцепочечной ДНК с нуклеотидной последовательностью,
комплементарной нуклеотидной последовательности молекулы мРНК (волнистая линия).
В результате синтеза получают пригодную для клонирования молекулу комплементарной ДНК (кДНК).
Для эффективной экспрессии структурному гену должны предшествовать нуклеотидные последовательности, обеспечивающие инициацию транскрипции (промотирующая последовательность) и начало трансляции (связывающий рибосому сайти стартовый кодон); за структурным геном должен следовать терминирующий кодон, означающий прекращение трансляции, и терминатор транскрипции (вспомните рис. 6.6). В векторы экспрессии входят все эти регуляторные последовательности, участок начала репликации, а также по меньшей мере один маркер селекции. С помощью клонирования и синтетическими методами получено большое число регулирующих экспрессию последовательностей, пригодных для включения в векторы экспрессии. Чаще других используются промоторы lac, trp и λρL.
Как правило, желательно, чтобы применяющийся промотор был регулируемым, т. е. включался только при каком-либо изменении параметров среды, например при добавлении индуктора, инактивации репрессора или при изменении температуры. В хорошо отработанной системе экспрессии содержание белка, отвечающего клонированному гену, может достигать 70% от всех клеточных белков, хотя обычно эта величина составляет от 10 до 25%. Поскольку продукт экспрессии не выполняет никакой полезной для клетки-хозяина функции, столь серьезное отвлечение биосинтезирующего аппарата клетки на синтез не нужных клетке белков может сказаться на скорости роста клеток и даже на их жизнеспособности. Поэтому в практической работе, как правило, условия культивирования в реакторе меняют таким образом, чтобы сначала экспрессия клонированного гена была практически подавлена и обеспечивался в основном быстрый рост клеточной массы вплоть до достижения необходимой плотности клеток, а затем с максимальной скоростью осуществлялась бы экспрессия.
Когда в Е. coli концентрация чужеродных белков достигает высокого уровня, они аккумулируются в клетке в виде плотных образований (рис. 6.27). Последние представляют собой главным образом чужеродный белок, но содержат и небольшие количества бактериальных белков, причем чужеродный белок обычно находится в денатурированном состоянии и клетки, содержащие плотные образования, часто имеют искаженную форму. Поэтому, хотя высокая концентрация белка в той или иной степени облегчает процессы выделения, его дальнейшая стабилизация и превращение в активную форму обусловливают необходимость дополнительных технологических стадий.
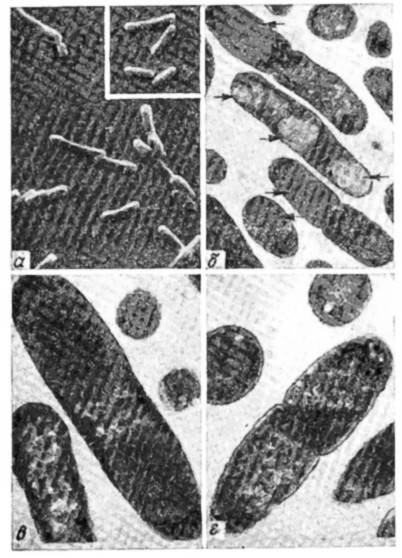
РИС. 6.27. а – электронная сканирующая микрофотография рекомбинантных клеток Е. coli,
продуцирующих химерный белок политриптофан-проинсулин. Рекомбинантные клетки
морфологически отличаются от нормальных клеток Е. coli (на вставке, Х5300)
удлиненной формой и наличием утолщений;
б – электронная просвечивающая микрофотография
рекомбинантных клеток Е. coli, продуцирующих химерный белок β-галактозидаза–цепь А инсулина (X17500);
в – рекомбинантные клетки Е. coli, продуцирующие политриптофан-проинсулин (Х30 000);
г–в клетках Е. coli, не содержащих плазмид, белковые включения отсутствуют (Х30 000).
Конечная продуктивность рекомбинантной культуры может лимитироваться обусловленной тем или иным фактором нестабильностью белка. Бактерия Е. coli содержит внутриклеточные протеазы, способные каким-то образом узнавать небольшие аномальные белки и разрушать их. В обычных клетках эти протеазы выполняют полезную функцию, разрушая ошибочно синтезированные белки до аминокислот, которые затем могут вновь использоваться для синтеза белков; в рекомбинантных системах подобные протеазы могут быстро расщепить весь чужеродный для клетки белок. Для повышения устойчивости последнего часто методами генной инженерии синтезируют гибридный белок, состоящий из белка Е. coli, к которому (как правило, к С-концу) присоединен чужеродный белок. Такой подход применялся для стабилизации ряда относительно небольших полипептидов, в том числе соматостатина (14 аминокислот), цепей А (21 аминокислота) и В (30 аминокислот) инсулина человека. В этих случаях роль соединительного звена между белковым фрагментом Е. coli и пептидом человека выполнял остаток метионина; при последующей обработке бромцианом, расщепляющим полипептидные цепи по остаткам метионина, образовывался активный пептид человека. (К счастью, ни один из перечисленных полипептидов не содержал остатков метионина!)
На рекомбинантных популяциях могут сказываться две различные формы генетической нестабильности. Во-первых, определенная часть вновь образующихся клеток может не содержать плазмид. В этом случае благодаря так называемой сегрегационной неустойчивости не несущие плазмид клетки могут подавлять рост содержащих плазмиды продуктивных клеток, если только система не находится под давлением отбора. Однако даже под давлением отбора сегрегационная нестабильность снижает общую скорость роста и продуктивность рекомбинантной популяции. Другой тип нестабильности – структурная неустойчивость – приводит к неспособности клеток синтезировать активный белок. Причина этой нестабильности может заключаться в мутации структурного гена или регуляторных нуклеотидных последовательностей. К сожалению, эта форма нестабильности не поддается регуляции путем давления отбора.
6.4.5. Генетическая инженерия с участием других клеток-хозяев
Е. coli – излюбленный объект подавляющего большинства работ по клонированию и генетической инженерии, поскольку этот организм наиболее детально изучен на молекулярном уровне. В то же время Е. coli практически не известна в микробиологической промышленности; будучи грамотрицательной бактерией, Е. coli содержит в своей наружной оболочке токсичные липополисахариды (заражение крови Е. coli приводит к летальному исходу в 50% случаев) и не секретирует белки в среду. К тому же как прокариота Е. coli не способна осуществлять характерные для эукариотических клеток сплайсинг и посттрансляционную модификацию белков. В силу этих и ряда других причин большой интерес представляет разработка систем клонирования и экспрессии в других, отличных от Е. coli организмах. При этом во всех случаях важно иметь в виду следующие факторы, которые могут препятствовать экспрессии чужеродного гена:
1. Разрушение чужеродных ДНК или РНК нуклеазами клеткихозяина.
2. Несрабатывание механизма репликации вектора.
3. Низкая активность промотора или терминатора транскрипции.
4. Неполный сплайсинг imPHK.
5. Неэффективная трансляция.
6. Гидролиз протеазами.
Кроме того, необходима эффективная методика, обеспечивающая введение вектора в клетку-хозяина с высоким выходом.
Известны методы клонирования и экспрессии генов в нескольких бактериях. Из последних наиболее хорошо изучена, по-видимому, Bacillus subtilis – грамположительный, непатогенный, непаразитический микроорганизм, использовавшийся до начала эры генетической инженерии в микробиологической промышленности для производства ряда ферментов и полипептидных антибиотиков. В. subtilis секретирует некоторые свои белки в среду. В генетической инженерии последнее свойство особенно удобно, поскольку секретируемые белки обычно не загрязнены большим количеством очень близких внутриклеточных белков. К тому же секреция белка в среду в принципе позволяет достичь более высокой концентрации синтезируемого белка по сравнению с внутриклеточными белками. (Каков верхний предел количества внутриклеточного белка на единицу объема культуральной жидкости?)
В В. subtilis можно клонировать ряд плазмид и бактериофагов. Для введения в В. subtilis чужеродной ДНК могут применяться методы трансформации, трансдукции и слияния протопластов. В В. subtilis удалось успешно осуществить экспрессию нескольких белков млекопитающих, в том числе инсулина и интерферонов, однако вплоть до момента написания этой главы извлечение чужеродных белков из В. subtilis оставалось проблематичным.
Технология клонирования применима и к ряду штаммов Pseudomonas и Streptomyces. Путем сочетания генетической инженерии с методами мутации и селекции удалось получить штаммы Pseudomonas с совершенно новым механизмом метаболизма; эти штаммы, например, могут расти на обычно токсичных хлорированных углеводородах как на единственном источнике углерода. Большое промышленное значение различных видов Streptomyces обусловило интенсивное изучение возможностей применения методов генетической инженерии к этим микроорганизмам. Целью этих работ является поиск штаммов, которые были бы более продуктивными в отношении имеющих практическое значение ферментов или синтезировали бы новые полусинтетические или гибридные антибиотики.
Последние примеры заслуживают несколько более детального обсуждения. Здесь мы впервые сталкиваемся с метаболической инженерией. В целом генетическая инженерия позволяет не только синтезировать белки, представляющие интерес как таковые, но и вводить в живую клетку особые ферменты, регуляторные белки, пермеазы и вообще любые другие белки. Таким путем мы можем придать клетке совершенно новую ферментативную, регуляторную или транспортную активность, найти которую среди нативных организмов или получить путем случайного мутагенеза было бы крайне маловероятно. Отсюда следует, что теперь мы в состоянии строго определенным и направленным путем перестраивать и реконструировать отдельные участки сети метаболических превращений клетки. В перспективе именно в этом направлении в основном и будет развиваться генетическая инженерия. Пока что достижению определенных результатов мешает недостаток данных о деталях конкретных важных путей метаболизма и о стадиях, лимитирующих скорость превращений по этим путям. К тому же наши знания о регуляции всей метаболической сети явно недостаточны для того, чтобы можно было попытаться предсказать влияние изменений в одном пути на ход других. Наконец, экспрессия большого числа новых генов в рекомбинантных клетках требует разработки новых приемов генетической инженерии.
В последние годы удалось добиться больших успехов в генетической инженерии эукариотических клеток. Особенно интенсивно изучались дрожжи Saccharomyces cerevisiae; этот микроорганизм выгодно отличается мощной генетической системой, способной к прямой экспрессии как некоторых эукариотических, так и ряда прокариотических генов, а также способностью трансформироваться с помощью чистой ДНК. Дрожжи также осуществляют по меньшей мере некоторые из типичных для эукариот посттрансляционных модификаций и секретируют некоторые белки в среду. Например, рекомбинантные S. eerevisiae продуцируют антиген австралийского гепатита В, который по степени гликозилирования и агрегации, по-видимому, не отличается от антигенов, обнаруженных в организме страдающих этим заболеванием людей. Генетически модифицированные дрожжи синтезируют и секретируют в среду иммунный интерферон человека (INF-γ).
Клонирование и экспрессия генов в клетках млекопитающих пока что осуществлялись только с помощью векторов на основе вируса SV40. Геном этого вируса (ковалентно замкнутая кольцевая ДНК) может реплицироваться в клетках млекопитающих как самостоятельно, так и после включения в хромосомы клетки-хозяина. Процесс экспрессии клонированного гена обеспечивается наличием в вирусе SV40 как промоторов, так и участка начала репликации, функционирующими в клетках млекопитающих.
Наиболее удобным для клонирования и идентификации ДНК остается все же быстрорастущий организм Е. coli, к которому применимо большинство методик генетической инженерии. Поэтому, если конечной целью являются многократная репликация вектора и экспрессия его генов в другом организме, часто оказывается удобным прибегнуть к помощи так называемого челночного вектора, который может реплицироваться как в Е. coli, так и в другом организме. Очевидно, что челночный вектор должен обладать двумя участками начала репликации – по одному на каждый организм-хозяин. На рис. 6.28 приведена схема челночного вектора, предназначенного для продуцирования иммунного интерферона (INF-γ) в клетках обезьян. В этом векторе фрагмент из 342 пар оснований, содержащий участок начала репликации и «поздний» промотор из SV40. связан с. фрагментом кДНК, в состав которой входят кодирующая последовательность npe-INF-γ, а также с фрагментом pBR322, включающим ген устойчивости к ампициллину и участок начала репликации Е. coli. Трансфекция (инфекция клеток чистой вирусной ДНК) трансформированных клеток обезьян линии COS-7 через 3–4 дня привела к INF-γ-активности порядка 50–100 единиц в 1 мл культуральной жидкости. Этот примерможет служить иллюстрацией тех преимуществ, которыми обладают генетически модифицированные клетки-хозяева млекопитающих в синтезе белков млекопитающих, когда все стадии биосинтеза белков и их секреции должны с максимально достижимой точностью воспроизводить соответствующие стадии, осуществляющиеся при синтезе и транспорте этих белков in vivo.
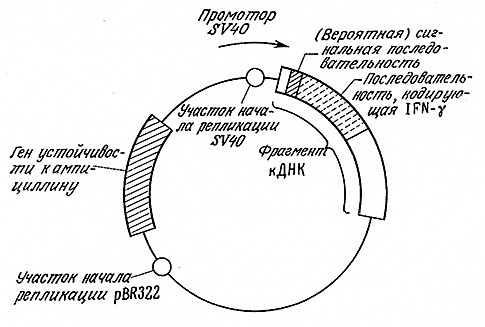
РИС. 6.28. Схема челночного вектора для Е. coli и клеток обезьяны, осуществляющего
экспрессию иммунного интерферона в клетках обезьяны и его секрецию.
Успешно развиваются также работы по введению чужеродных генов в целые организмы растений и животных и их последующей экспрессии. Хотя анализ методологии этих работ не входит в тематику настоящей книги, их потенциальная значимость для человека очевидна. Открывающие новые перспективы научные и технические достижения в то же время стимулировали широкие дискуссии (в том числе и в литературе) о возможности неблагоприятного воздействия биотехнологии на общество [24].
6.4.6. Заключение
Приведенный в табл. 6.8 перечень основных этапов синтеза белка с помощью рекомбинантных ДНК может служить кратким обзором темы настоящего раздела. Запомните, что клонирование необходимого гена и его экспрессия эффективно выполняются как отдельные последовательные операции. Следует еще раз подчеркнуть необходимость постоянного контроля любых превращений ДНК путем определения нуклеотидной последовательности и изучения фрагментов, образующихся при расщеплении рестриктазами.
Таблица 6.8. Последовательность этапов работы по клонированию гена и экспрессии соответствующего белка методами технологии рекомбинантных ДНК

Принимая во внимание ту большую роль, которую будет играть технология рекомбинантных ДНК в будущем, технологу-биохимику необходимо значительно глубже изучить основы молекулярной биологии и соответствующей методологии. Знание этих отраслей науки необходимо при знакомстве с современной биологической литературой, а также при разработке в сотрудничестве с молекулярными биологами оптимальных организмов и процессов. Факторы, определяющие продуктивность рекомбинантных популяций (уровни экспрессии гена, активности внутриклеточных протеаз, генетическая стабильность и т. п.), зависят от нуклеотидной последовательности вектора, генетических свойств клетки-хозяина, состава среды, конструкции биореактора и условий его работы. Для понимания этих зависимостей, а также различных взаимосвязей в генетически видоизмененном организме необходимы совместные усилия специалистов в области биохимической технологии и молекулярной биологии.
