Технология рекомбинантных ДНК (ее называют также молекулярным клонированием или генной инженерией) – это совокупность экспериментальных процедур, позволяющая осуществлять перенос генетического материала (дезоксирибонуклеиновой кислоты, ДНК) из одного организма в другой. Никакого единого, универсального набора методик здесь не существует, но чаще всего эксперименты с рекомбинантной ДНК проводят по следующей схеме (рис. 4.1).
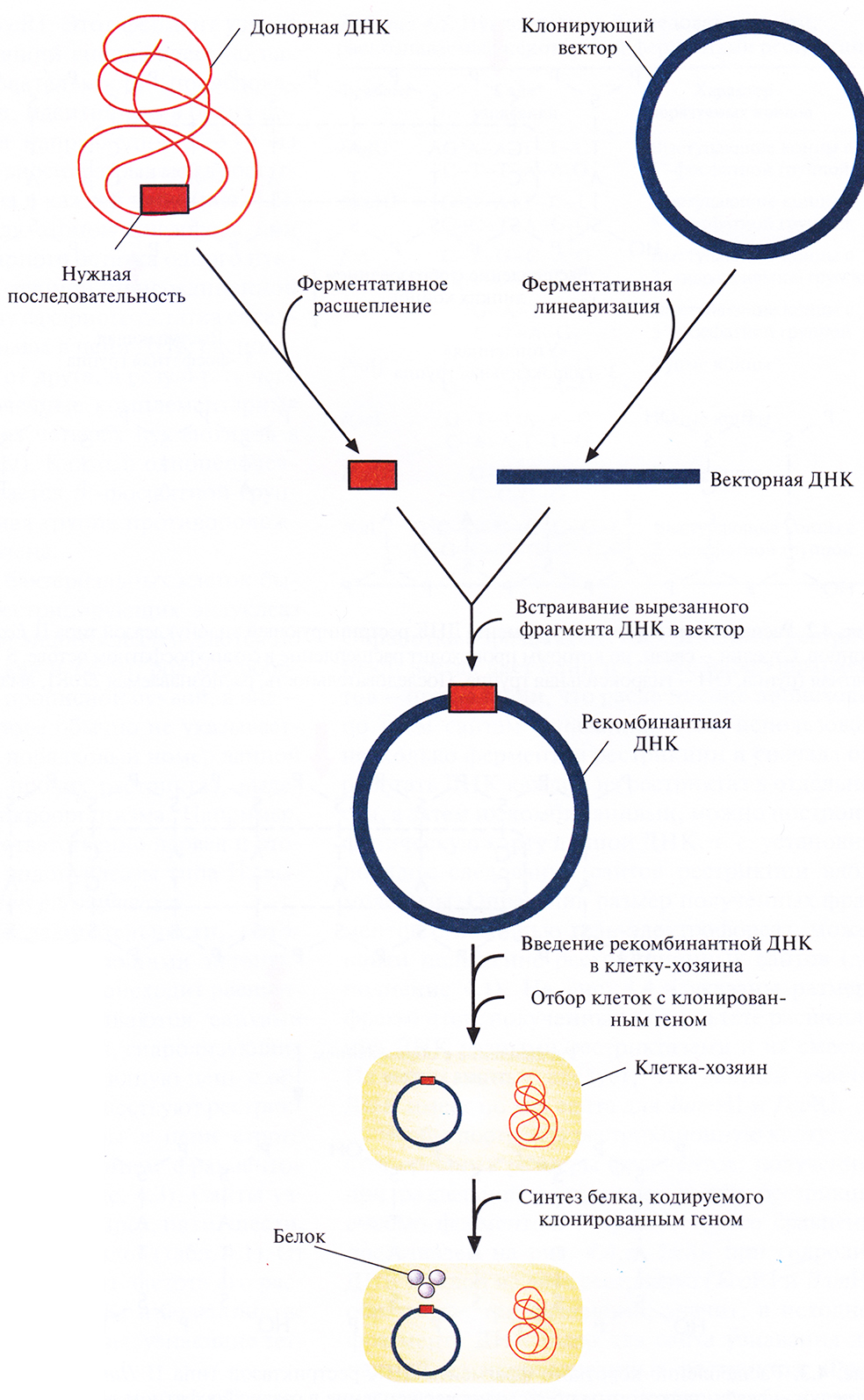
Рис. 4.1. Клонирование рекомбинантной ДНК. Донорную ДНК расщепляют рестрицируюгцей эндонуклеазой и встраивают в клонирующий вектор. Полученную конструкцию вводят в популяцию клеток-хозяев, идентифицируют те клетки, которые содержат рекомбинантную ДНК, и культивируют их. При необходимости можно индуцировать экспрессию клонированного гена в клетках-хозяевах и получить кодируемый им белок.
- Из организма – донора нужных генов – экстрагируют нативную ДНК (клонируемая ДНК, встраиваемая ДНК, ДНК-мишень, чужеродная ДНК), подвергают ее ферментативному гидролизу (расщепляют, разрезают) и соединяют (лигируют, сшивают) с другой ДНК (вектор для клонирования, клонирующий вектор) с образованием новой, рекомбинантной молекулы (конструкция «клонирующий вектор–встроенная ДНК»).
- Эту конструкцию вводят в клетку-хозяина (реципиент), где она реплицируется и передается потомкам. Этот процесс называется трансформацией.
- Идентифицируют и отбирают клетки, несущие рекомбинантную ДНК (трансформированные клетки).
- Получают специфический белковый продукт, синтезированный клетками-хозяевами, что служит подтверждением клонирования искомого гена.
Предпосылками к созданию технологии рекомбинантных ДНК послужили многие открытия в области молекулярной биологии, энзимологии нуклеиновых кислот и молекулярной генетики бактериальных вирусов и внехромосомных элементов бактерий (плазмид). Конструирование рекомбинантных молекул осуществляется с помощью целого арсенала ферментов – обязательного и незаменимого инструмента практически всех этапов этого сложнейшего процесса. Речь идет прежде всего о ферментах рестрикции (рестрицирующих эндонуклеазах, рестриктазах), которые узнают и расщепляют специфические нуклеотидные последовательности в двухцепочечной молекуле ДНК.
РЕСТРИЦИРУЮЩИЕ ЭНДОНУКЛЕАЗЫ
При молекулярном клонировании важно, чтобы расщепление донорной и векторной ДНК происходило в строго определенных участках (сайтах) с образованием дискретного и воспроизводимого набора фрагментов. Если пропустить хромосомную ДНК через шприц с иглой малого диаметра или обработать ее ультразвуком, то мы получим фрагменты длиной от 0,3 до 5 т.п.н. К сожалению, в ходе этих простых операций разрывы двухцепочечных молекул происходят случайным образом, так что при каждой обработке препарата ДНК получается совершенно новый набор фрагментов. Молекулярное клонирование стало возможным только после выделения высокоспецифичных бактериальных ферментов, которые узнают определенные последовательности оснований в двухцепочечной молекуле ДНК и расщепляют обе цепи. Эти ферменты называются рестрицирующими эндонуклеазами типа II.
Одна из первых рестрицирующих эндонуклеаз типа II была выделена из бактерии Escherichia coli и получила название EcoRl. Этот фермент узнает участок ДНК, содержащий специфическую палиндромную последовательность (последовательность-перевертыш, идентичную в обеих цепях при прочтении в направлении 5'→3') из шести пар оснований и вносит разрыв между остатками гуанина и аденина в каждой цепи (рис. 4.2), расщепляя связь между атомом кислорода при 3'-атоме углерода сахарного остатка одного нуклеотида и фосфатной группой, присоединенной к 5 '-углеродному атому сахарного остатка соседнего нуклеотида. Разрывы в цепи ДНК располагаются наискось друг от друга, в результате чего образуются одноцепочечные комплементарные концы с «хвостами» из четырех нуклеотидов в каждом (липкие концы). Каждый одноцепочечный «хвост» заканчивается 5'-фосфатной группой, а З'-гидроксильная группа противоположной цепи как бы утоплена.
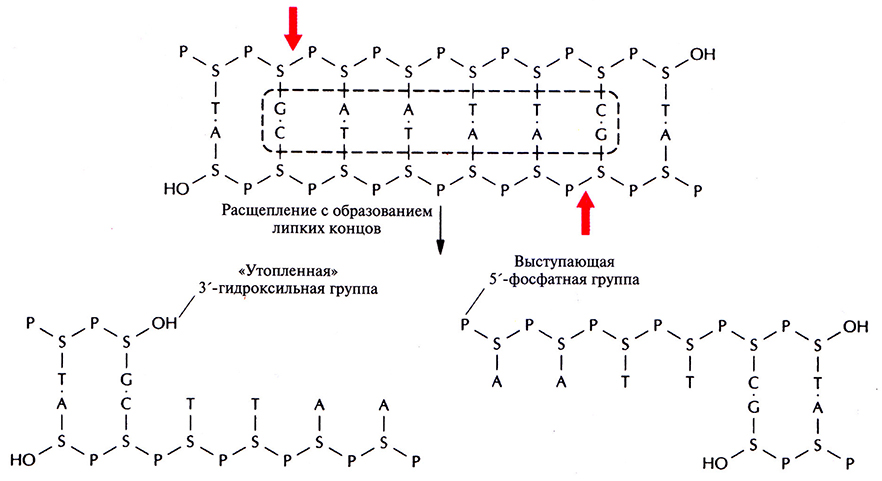
Рис. 4.2. Расщепление короткого фрагмента ДНК рестрицирующей эндонуклеазой типа II EcoRI с образованием липких концов. Стрелки – связи, по которым происходит расщепление в сахарофосфатном остове. S – дезоксирибоза, Р – фосфатная группа, ОН – гидроксильная группа. Последовательность, распознаваемая EcoRI, выделена штриховой линией.
Помимо EcoRI, из бактериальных клеток были получены сотни рестрицирующих эндуклеаз типа II. Названия этим эндонуклеазам даются по такому же принципу, как и EcoRI. род микроорганизма обозначается прописной буквой, а вид – двумя строчными, штамм обычно не указывается. Римские цифры – порядковый номер данной эндонуклеазы в ряду прочих рестриктаз, выделенных из данного микроорганизма. Например, Hpal и НраII – это соответственно первая и вторая рестрицирующие эндонуклеазы типа II, выделенные из Haemophilus parainfluenzae.
Палиндромные последовательности, которые распознаются рестрицирующими эндонуклеазами тина II и в которых происходит расщепление молекулы ДНК, называются сайтами узнавания. Помимо рестриктаз, гидролизующих (расщепляющих) полинуклеотидную цепь с образованием липких концов, существуют рестриктазы, которые вносят разрывы в цепи строго друг против друга с образованием фрагментов ДНК с «тупыми» концами (рис. 4.3). Сайты узнавания могут состоять из четырех, пяти, шести, восьми или более пар нуклеотидов (табл. 4.1). От длины сайта узнавания зависит частота его распространения в молекуле ДНК; в большинстве случаев используют рестриктазы, узнающие тетра- и гексануклеотиды.
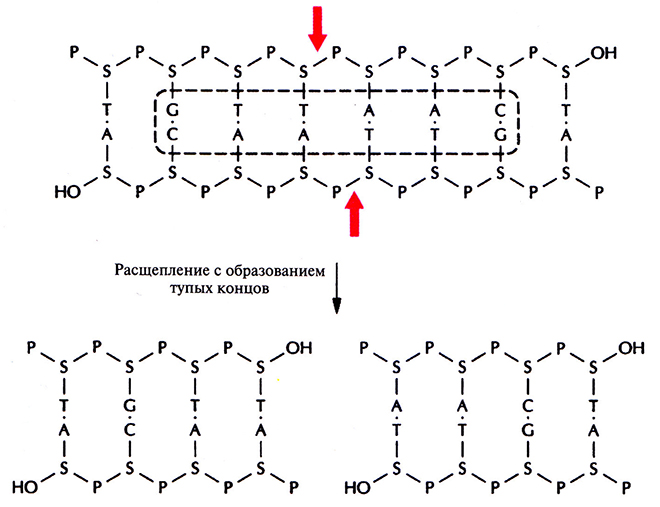
Рис. 4.3. Расщепление короткого фрагмента ДНК рестриктазой типа II HindII с образованием тупых концов Стрелки – связи, по которым происходит расщепление в сахарофосфатном остове. Буквенные обозначения – те же, что и на рис. 4.2. Последовательность, распознаваемая рестриктазой HindII, выделена штриховой линией.
Таблица 4.1. Нуклеотидные последовательности,
распознаваемые некоторыми ферментами рестрикции

Рестрицирующие эндонуклеазы типа II играют ключевую роль при генном клонировании.
Обработка образца ДНК определенной рестриктазой всегда дает один и тот же набор фрагментов – при условии, что расщепление происходит по всем сайтам узнавания. Если использовать несколько ферментов рестрикции и сначала обработать ДНК каждой из рестриктаз в отдельности, а затем их комбинациями, можно построить физическую карту данной ДНК, т. е. установить порядок следования сайтов рестрикции вдоль молекулы. Определив размер полученных фрагментов с помощью гель-электрофореза, можно найти положение рестрикционных сайтов (дополнение 4.1). На рис. 4.4,А указаны размеры фрагментов, полученных в результате расщепления ДНК разными рестриктазами и их смесью. Из этих данных следует, что данный участок ДНК имеет по два сайта для BamHl и ЕсоRI.
ДОПОЛНЕНИЕ 4.1
Гель-электрофорез
Для разделения белков и нуклеиновых кислот широко применяется метод гель-электрофореза. Его принцип заключается в следующем. Исследуемый препарат (раствор белка, ДНК или РНК) вносят в лунку, расположенную у края геля – полужидкой среды с сетчатой пространственной структурой (обычно для электрофореза используют тонкие пластины геля). Находящиеся в буферном растворе макромолекулы обладают некоторым суммарным электрическим зарядом, и когда через гель пропускают электрический ток, они перемещаются в электрическом поле. Молекулы одинакового размера (и одинакового заряда) движутся единым фронтом, образуя в геле дискретные невидимые полосы. Чем меньше размер молекул, тем быстрее они движутся. Постепенно исходный препарат, состоящий из разных макромолекул, разделяется на зоны, распределенные по длине пластинки.
За ходом электрофореза следят по перемещению в геле красителя – заряженного низкомолекулярного вещества, которое вносят в каждую лунку перед началом электрофореза. Когда краситель достигает конца пластины, электрофорез останавливают, а гель окрашивают красителем, прочно связывающимся с белками или нуклеиновыми кислотами. Если образец представляет собой дискретный набор макромолекул разного размера, то после электрофореза получается набор четких полос, расположенных одна под другой. Если же распределение молекул по размеру более или менее непрерывно, то получается смазанная картина. По интенсивности окраски полос можно судить о концентрации макромолекул в образце.
Чтобы определить относительную молекулярную массу разделенных фрагментов, одновременно проводят электрофорез маркерных макромолекул с известными молекулярными массами. Набор маркеров должен охватывать весь диапазон молекулярных масс в данной системе. Образец маркерных молекул вносят в отдельную лунку, расположенную вблизи одного из краев пластинки (или в две лунки у двух разных краев). Логарифм относительной молекулярной массы маркера линейно связан с его электрофоретической подвижностью Rf – величиной, равной отношению расстояний, пройденных маркерной молекулой и красителем (фронтом растворителя). Построив график зависимости логарифма относительных молекулярных масс маркеров от Rf, можно найти относительную молекулярную массу каждого компонента образца. Относительная мол. масса белков измеряется в дальтонах, двухцепочечных нуклеиновых кислот – в числе пар нуклеотидов, одноцепочечных – в числе нуклеотидов.
Для разделения белков обычно используют полиакриламидный гель (ПААГ). Он образуется при сополимеризации акриламида и бисакриламида, использующегося в качестве сшивки линейных полимеров акриламида. Размер ячеек в полиакриламидной «сетке» зависит от концентрации акриламида и соотношения между количеством акриламида и бисакриламида. Белок перед электрофорезом часто обрабатывают анионным детергентом додецилсульфатом натрия (ДСН), что позволяет проводить фракционирование в зависимости только от одного параметра – молекулярной массы, а зависимость от конформации, плотности упаковки полипептидной цепи и др. исключается. Электрофорез в ПААГ-ДСН позволяет разделять белки с мол. массой от 20 до 200 кДа.
Для электрофоретического разделения нуклеиновых кислот среднего размера обычно применяют агарозные гели. Агароза – это особо чистая фракция, получаемая из агара или непосредственно из агарообразующих морских водорослей. В 1,0% агарозном геле можно разделять молекулы ДНК размером от 600 до 20 000 п. н. Для фракционирования более крупных молекул ДНК (миллионы пар оснований), денатурированной ДНК и РНК приходится использовать специальные системы электрофореза. Иногда для решения специальных задач для разделения ДНК применяют полиакриламидные гели. Так, в 20% полиакриламидном геле можно разделить фрагменты ДНК, состоящие всего из шести оснований и различающиеся лишь одним нуклеотидом.

Рис. 4.4. Картирование сайтов рестрикции. А. Результаты гель-электрофореза фрагментов ДНК, полученных ее расщеплением указанными ферментами. Очищенную ДНК гидролизовали рестриктазами ЕсоRI и ВатНI раздельно, а затем их смесью, проводили гель-электрофорез и визуализировали продукты окрашиванием бромистым этидием. Числа слева от горизонтальных полос – длина фрагментов в парах оснований. Б. Рестрикционная карта, построенная по электрофоретическим данным. Числа – расстояние между сайтами узнавания соответствующих ферментов.
Чтобы построить рестрикционную карту, следует сравнить размеры фрагментов, полученных при раздельной рестрикции и при рестрикции смесью ферментов. Результат такого сравнения представлен на рис. 4.4,Б. Если при гидролизе ДНК каждой из двух рестриктаз (ЕсоRI и ВатHI) образуются три фрагмента, значит, в исходном фрагменте ДНК было два сайта узнавания для каждой из использованных рестриктаз. Фрагмент размером 300 п. н., который образуется в результате гидролиза ЕсоRI, не расщепляется при гидролизе смесью ЕсоRI и ВатHI в отличие от ЕсоRI-фрагментов размером 850 и 500 п. н. Значит, два ЕсоRI-сайта находятся на расстоянии 300 п. н. друг от друга и между ними нет ВатHI-сайта, а в ЕсоRI-фрагментах длиной 850 и 500 п.н. есть по одному BamHl-сайту. Фрагмент размером 950 п. н., который образуется при обработке ДНК рестриктазой ВатHI, при двойном гидролизе расщепляется ЕсоRI на три фрагмента (250+300+400 = 950 п. н.). Значит, два ВатHI-сайта находятся на расстоянии 250 и 400 п. н. по разные стороны от сайтов для ЕсоRI. ВатHI расщепляет ЕсоRI-фрагмент длиной 850 п. н. на фрагменты длиной 250 и 600 п. н., а один из сайтов для ЕсоRI находится на расстоянии 250 п. н. от сайта для ВатHI, значит, фрагмент 600 п. н. должен содержать один из концов исходной молекулы ДНК. Далее, мы видим, что ВатHI расщепляет ЕсоRI-фрагмент длиной 500 п. н. на два фрагмента размером 100 и 400 п. н. и что один из ЕсоRI-сайтов отделен от ВатHI-сайта 400 п. н.; значит, фрагмент длиной 100 п. н. должен содержать другой конец исходной молекулы. Карта на рис. 4.4, Б иллюстрирует четкое соответствие между положением сайтов рестрикции и размерами фрагментов, получающихся при каждом гидролизе.
Расщепление рестрицирующими эндонуклеазами имеет еще одно применение. Когда два разных образца ДНК обрабатывают одной и той же рестриктазой с образованием фрагментов с липкими концами, а затем смешивают эти образцы, то благодаря комплементарному спариванию липких концов фрагментов разных образцов могут образовываться новые комбинации генов – рекомбинантные ДНК (рис. 4.5).
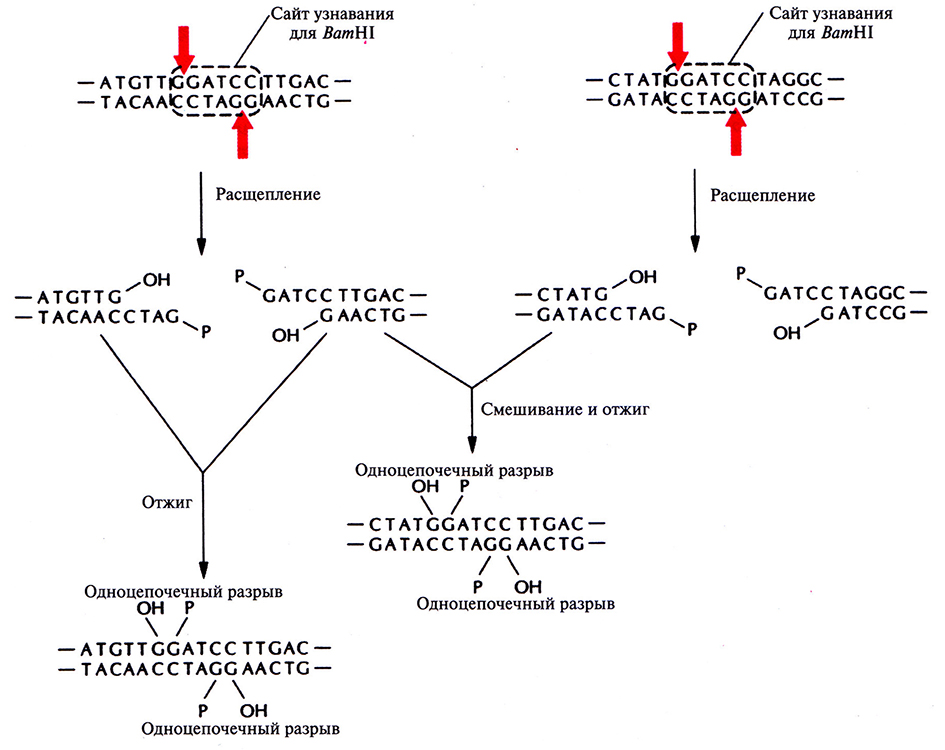
Рис. 4.5. Отжиг комплементарных липких концов фрагментов, образующихся при расщеплении двух разных образцов ДНК рестрицирующей эндонуклеазой ВатHI. Четыре фрагмента, представленных на рисунке, могут соединиться друг с другом с образованием шести разных молекул ДНК (на рисунке показаны не все возможные комбинации). Фрагменты удерживаются вместе водородными связями, образующимися между четырьмя основаниями липких концов, но эти связи недостаточно прочны, чтобы молекулы в растворе оставались стабильными длительное время.
Для осуществления молекулярного клонирования недостаточно одних только ферментов рестрикции. Во-первых, водородные связи между теми четырьмя основаниями, которые образуют липкие концы, недостаточно прочны, чтобы удержать два объединившихся фрагмента ДНК. Необходим какой-то инструмент для устранения разрыва в сахарофосфатном остове молекулы, т. е. для восстановления связи между З'-гидроксильной концевой группой одной цепи и 5'-фосфатной группой другой. Таким инструментом является ДНК-лигаза бактериофага Т4. Этот фермент катализирует образование фосфодиэфирных связей между концами полинуклеотидных цепей, которые уже удерживаются вместе благодаря спариванию липких концов. Кроме того, ДНК-лигаза Т4 «сшивает» тупые концы, которые сближаются друг с другом после того, как объединяемые фрагменты связываются с ферментом (рис. 4.6). Во-вторых, объединение разных молекул ДНК само по себе бесполезно, если вновь образованные комбинации (рекомбинантные ДНК) не будут реплицироваться в клетке-хозяине. Таким образом, если одна часть рекомбинантной молекулы ДНК несет нужный ген, который предполагается клонировать, то другая должна содержать информацию, необходимую для репликации в клетке рекомбинантной ДНК. Чтобы решить эту проблему, используют клонирующие векторы. В-третьих, при рестрикции ДНК образуется смесь разнообразных фрагментов, и после их дотирования с векторной ДНК образуется множество различных комбинаций. Необходимо уметь распознавать те реципиентные клетки, которые содержат ДНК с нужной нуклеотидной последовательностью. Для этого используют различные системы скрининга.
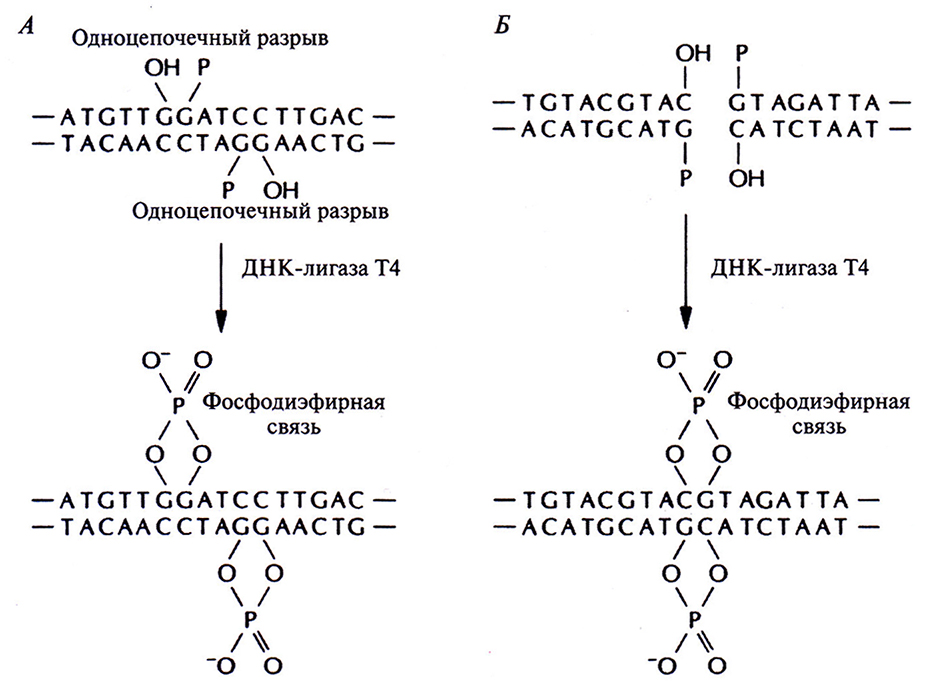
Рис. 4.6. ДНК-лигаза Т4 образует фосфодиэфирные связи между 5'-фосфатными и 3 -гидроксильными группами в месте разрыва в остове двухцепочечной ДНК. А. Нитрование липких концов. Б Лигирование тупых концов.
ПЛАЗМИДНЫЕ ВЕКТОРЫ
Плазмиды – это внехромосомные автономно реплицирующиеся двухцепочечные кольцевые молекулы ДНК. Плазмиды есть практически у всех бактерий. Одни из них содержат информацию, обеспечивающую их собственный перенос из одной клетки в другую (F-плазмиды), другие несут гены устойчивости к антибиотикам (R-плазмиды) или специфические наборы генов, ответственных за утилизацию необычных метаболитов (плазмиды деградации). Есть плазмиды, в которых не обнаружены гены, выполняющие какие-то определенные функции (критические плазмиды; от англ. cryptic – скрытый, латентный). Размеры плазмид варьируют от менее 1 до более 5С0 т.п.н. Каждая из них содержит сайт начала репликации (ori), без которого репликация плазмиды в клетке-хозяине была бы невозможна.
Некоторые плазмиды представлены в клетке 10-100 копиями; они называются высококопийными Низкокопийные плазмиды присутствуют в клетке в числе 1–4 копий. На долю плазмидной ДНК обычно приходится 0,1–5,0% суммарной клеточной ДНК. Если две или более плазмиды не могут сосуществовать в одной и той же клетке, то говорят, что они принадлежат к одной группе несовместимости. Плазмиды, относящиеся к разным группам несовместимости, беспрепятственно существуют в одной клетке, независимо от числа копий. У некоторых микроорганизмов в одной клетке было обнаружено до 8–10 разных плазмид, при этом каждая из них выполняла свои функции, была представлена характерным для нее числом копий и относилась к своей собственной группе несовместимости. Одни плазмиды несут специфичный сайт инициации репликации и могут реплицироваться только в клетках одного вида. У других плазмид этот сайт менее специфичен, и они реплицируются в самых разных бактериальных клетках. Соответственно различают плазмиды с узким и с широким спектром хозяев.
Как автономно реплицирующиеся генетические элементы плазмиды обладают всеми основными свойствами, которые позволяют использовать их в качестве вектора для переноса клонируемой ДНК. Но довольно часто природные (немодифицированные, несконструированные) плазмиды бывают лишены некоторых обязательных для «высококачественного» вектора свойств. К таким важным свойствам относятся:
1. небольшой размер, поскольку эффективность переноса экзогенной ДНК в Е. coli значительно снижается при длине плазмиды более 15 т. п. н.;
2. наличие уникального сайта рестрикции, в который может быть осуществлена вставка;
3. наличие одного или более селективных генетических маркеров для идентификации реципиентных клеток, несущих рекомбинантную ДНК. Поэтому плазмидные векторы приходится создавать с помощью генной инженерии.
Плазмидный вектор pBR322
В 80-е годы плазмидный вектор pBR322 был одним из самых популярных универсальных векторов. Обычно обозначение плазмидного вектора включает строчную букву р (от англ. plasmid) и еще несколько букв, имеющих отношение к описанию вектора или к истории его создания. Так, буквы BR в обозначении плазмиды pBR322 указывают на авторство Ф. Боливара и Р. Родригеса, сконструировавших эту плазмиду, а число 322 – цифровое обозначение, взятое из их исследовательских протоколов. Длина плазмиды pBR322 – 4361 п. н. Она несет два гена устойчивости к антибиотикам (рис. 4.7), ампициллину (Аmpr) и тетрациклину (Tetr), а также уникальные сайты для ВатНI, HindIll и Sail в гене Tetr, один PstI-сайт в гене Ampr, один сайт для EcoRI, находящийся за пределами кодирующих последовательностей, и сигнал начала репликации, обеспечивающий репликацию исключительно в Е. coli. Плазмида реплицируется с образованием большого числа копий, в другие бактериальные клетки переносится с трудом.
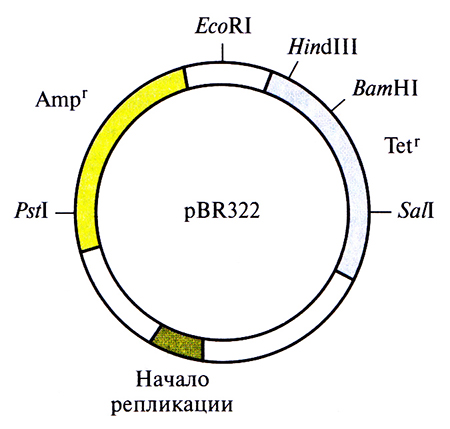
Рис. 4.7. Генетическая карта плазмидного вектора pBR322. Гены устойчивости к тетрациклину (Tetr) и ампициллину (Ampr) содержат уникальные сайты узнавания для HindIII, Sail, ВатHI и Pstl. EcoRI-сайт расположен вне этих генов. Длина вектора – 4361 п. н.
Как работает клонирующий вектор pBR322? Если очищенную кольцевую плазмиду pBR322 обработать рестриктазой, расщепляющей ее в единственном сайте, расположенном в одном из генов устойчивости к тому или другому антибиотику, то образуется линейная молекула с липкими концами. Такие молекулы смешивают с донорной ДНК, содержащей нужный ген и предварительно обработанной такой же рестриктазой. Поскольку липкие концы этих двух ДНК взаимно комплементарны, они спариваются с образованием гибридных молекул. Далее смесь обрабатывают ДНК-лигазой фага Т4 в присутствии АТР, в результате чего образуется множество разных комбинаций фрагментов, а также нежелательные продукты, в частности объединившиеся между собой фрагменты донорной ДНК и исходные плазмидные ДНК. Чтобы уменьшить количество последних, обрабатывают рестрицированную плазмидную ДНК щелочной фосфатазой, отщепляющей от линеаризованной молекулы 5'-фосфатные группы: ДНК-лигаза не может сшить концы дефосфорилированной линейной плазмидной ДНК (рис. 4.8). Что касается собственно рекомбинантных молекул ДНК, то хотя в них и имеются два одноцепочечных разрыва, ее фрагменты удерживаются вместе двумя фосфодиэфирными связями, образовавшимися с помощью ДНК-лигазы между дефосфорилированной плазмидной ДНК и рестрицированной донорной ДНК (рис. 4.8). После репликации в трансформированной клетке одноцепочечные разрывы устраняются системой лигирования клетки-хозяина.
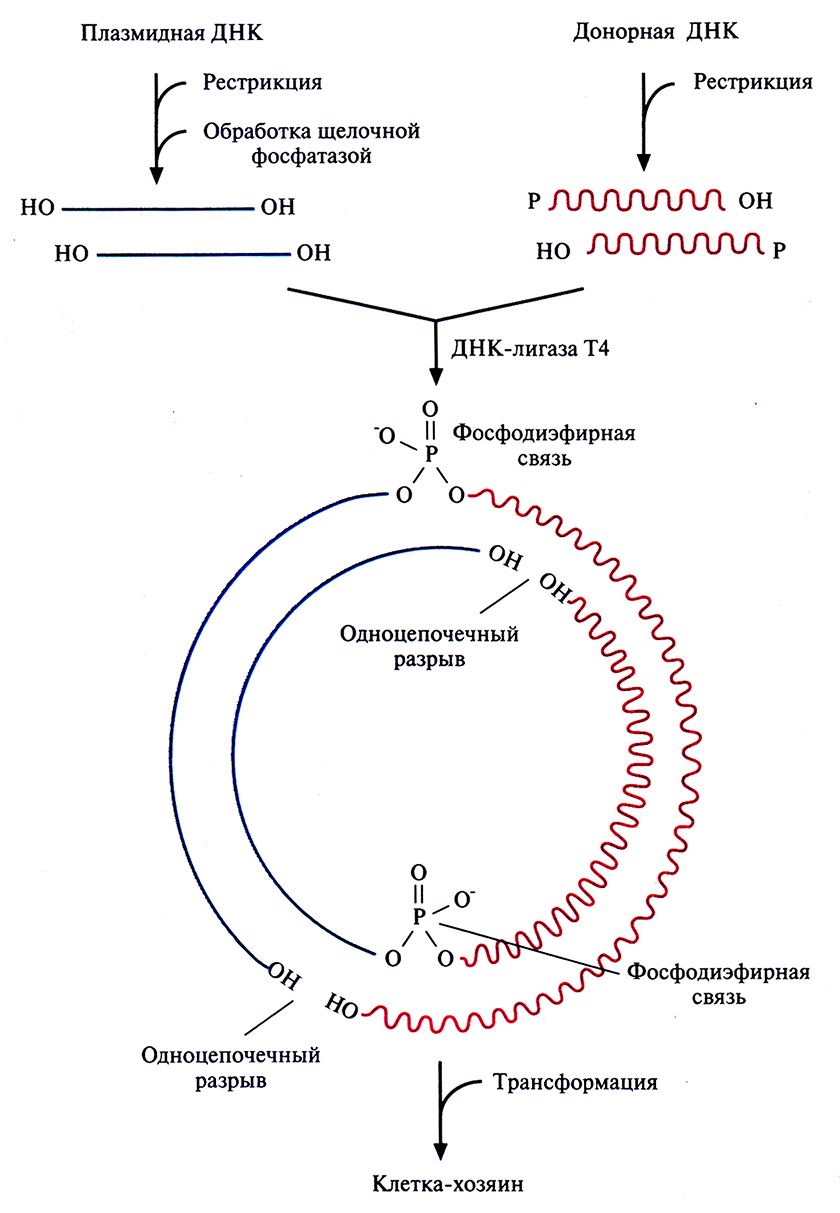
Рис. 4.8. Встраивание чужеродной ДНК в плазмидный вектор. Плазмидную ДНК, обработанную рестриктазой и щелочной фосфатазой, смешивают с рестрицированной донорной ДНК, содержащей нужный ген, и добавляют ДНК-лигазу. Два из четырех одноцепочечных разрыва при этом устраняются, и конструкция оказывается стабильной благодаря образовавшимся фосфодиэфирным связям. После введения гибридной ДНК в клетку-хозяина происходит ее репликация и образуются новые кольцевые молекулы уже без разрывов.
Трансформация и отбор
Теперь необходимо ввести рекомбинантную ДНК в клетку-хозяина. Этот процесс называется трансформацией. Для его осуществления используют специально разработанные приемы, например подвергают клетки высокотемпературному воздействию и обрабатывают их хлористым кальцием (СаС12). Однако эффективность трансформации все же остается невысокой, обычно трансформируется не более одной клетки из тысячи. Таким образом, большинство клеток после проведения трансформации не содержат рекомбинантной ДНК. В некоторых из них появляется воссоединившаяся кольцевая плазмидная ДНК, избежавшая дефосфорилирования щелочной фосфатазой, в других – неплазмидная ДНК и лишь в некоторых – плазмида со встроенным фрагментом чужеродной ДНК (гибридная плазмида).
Как мы уже говорили, внехромосомная ДНК, не содержащая точки начала репликации, не может реплицироваться в бактериальной клетке. Таким образом, проникновение в клетку экзогенной ДНК еще не означает, что она будет поддерживаться в хозяйской клетке. Далее, для сохранения рекомбинантной ДНК в клетке-хозяине в первоначальном виде необходимо, чтобы в клетке отсутствовали гены, кодирующие синтез рестриктаз, которые могут привести к ее деградации, и чтобы клетка имела фенотип RecA– (такие клетки неспособны к общей рекомбинации, так что экзогенная ДНК не будет модифицироваться в результате гомологичной рекомбинации).
Затем необходимо идентифицировать клетки, содержащие рекомбинантную ДНК. Способ идентификации должен быть как можно более простым, поскольку приходится проверять огромное число клеток. В системе pBR322, в которой чужеродная ДНК встраивается в сайт ВатHI, специфическая идентификация состоит из двух этапов. Сначала клетки после трансформации высевают на питательную среду, содержащую ампициллин. В таких условиях могут вырасти только те клетки, в которых присутствует интактный ген Ampr – или в составе интактной плазмиды pBR322, или в составе гибридной плазмиды; трансформированные клетки чувствительны к ампициллину. Сайт BamHI расположен в гене Tetr плазмиды pBR322 (рис. 4.7); встраивание в этот ген фрагмента ДНК прерывает кодирующую последовательность, и устойчивость к тетрациклину утрачивается. Таким образом, клетки, несущие гибридную плазмиду, устойчивы к ампициллину, но чувствительны к тетрациклину, а клетки, получившие интактную плазмиду pBR322, несут ген Tetr и устойчивы как к ампициллину, так и к тетрациклину.
На втором этапе проводят разделение этих двух вариантов. Клетки, выросшие на среде с ампициллином, переносят на среду с тетрациклином методом перепечатки. Клетки, образующие колонии на чашках с тетрациклином, содержат интактную плазмиду pBR322, поскольку, как мы уже говорили, они устойчивы и к ампициллину, и к тетрациклину. Клетки, не выросшие на чашках с тетрациклином, чувствительны к этому антибиотику, значит, они содержат гибридную плазмиду pBR322.
Среди колоний, выросших на среде с ампициллином, выделяют те, которые оказались чувствительны к тетрациклину, и из каждой колонии получают индивидуальные клеточные клоны или (чаще делают именно так) объединяют все колонии, устойчивые к ампициллину и чувствительные к тетрациклину, и культивируют их вместе. Далее можно провести дополнительный скрининг и идентифицировать те клетки, которые несут гибридную плазмиду pBR322 со специфической вставкой. Присутствие сайтов Hindlll и Sail в гене Tetr и сайта Pstl в гене Ampr плазмиды pBR322 позволяет изменить локализацию клонированных фрагментов чужеродной ДНК. Если для встраивания используется сайт Pstl, то отбор проводится по той же схеме, но в другом порядке, т. е. сначала высевают клетки на среду с тетрациклином, а затем – с ампициллином.
Другие плазмидные векторы
Идея использовать pBR322 как вектор для клонирования была вполне удачной, но эта плазмида содержит лишь несколько сайтов рестрикции, а отбор трансформированных клеток занимает много времени. Это привело к необходимости разработки альтернативных систем клонирования. Например, плазмида pU С19 длиной 2686 п. н. содержит: ген устойчивости к ампициллину; регулируемый сегмент гена р-галактозидазы (lacZ') лактозного оперона Е. coir, ген lacl, кодирующии репрессор, который контролирует экспрессию гена lacZ'; полилинкер – короткую последовательность с множеством уникальных сайтов узнавания для эндонуклеаз (ЭсoRI, Sacl, Kpnl, Xmal, Smal, BamHl, Xbal, Sall, Hincll, Accl, BspMl, Pstl, Sphl и Hindlll);, точку начала репликации плазмиды pBR322 (рис. 4.9).
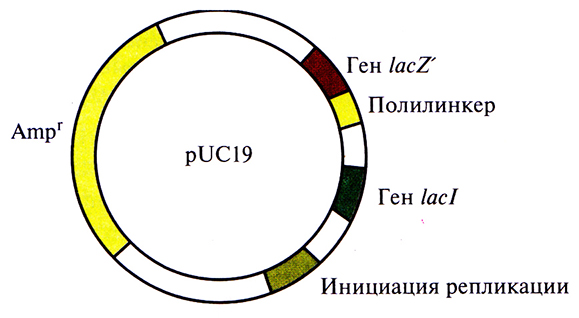
Рис. 4.9. Генетическая карта плазмидного вектора pUC19. Плазмида состоит из 2686 пар нуклеотидов и содержит уникальные сайты узнавания для EcoRI, Sacl, Kpnl, Xmal, Smal, ВатHl, Xbal, Sail, Hincll, Accl, Pstl, BspMl, Sphl и Hindlll, локализованные в полилинкере; ген устойчивости к ампициллину; сайт инициации репликации, функционирующий в Е. coli; ген lacl, контролирующий синтез репрессора, который блокирует транскрипцию гена lacZ' в отсутствие индуктора ИПТГ.
ВАЖНАЯ ВЕХА
При расщеплении ДНК рестриктазой RI образуются фрагменты с липкими концами
J.E. Mertz, R.W. Davis Proc. Natl. Acad. Set. USA 69: 3370-3374, 1972
Технология рекомбинантных ДНК включает создание вектора – «переносчика» клонируемой ДНК, специфическое встраивание в него этой ДНК с образованием химерной конструкции, введение конструкции в клетку-хозяина и идентификацию клеток, несущих рекомбинантную ДНК. Незаменимым рабочим инструментом в этих манипуляциях являются рестрицирующие эндонуклеазы типа II. Их применяют как при создании векторов (см., например, Bolivar et al., Gene 2: 95–113, 1977), так и при встраивании в них нужных генов. В 1968 г. М. Мезелсон и др. (Nature 217: 1110–1114) показали, что способность одного из штаммов Е. coli сдерживагь размножение инфицирующего его вируса (бактериофага) обусловливается наличием в клетке фермента, который расщепляет фаговую ДНК. Позже Мерц и Дэвис установили, что этот фермент, рестриктаза RI (получившая название EcoRl), расщепляет молекулу ДНК в специфическом сайте с образованием комплементарных («липких») концов. Линейные молекулы, образовавшиеся при расщеплении кольцевой ДНК рестриктазой EcoRl, часто вновь замыкаются в кольцо благодаря спариванию комплементарных концов. Объединившиеся фрагменты удерживаются вместе водородными связями; их концы можно ковалентно сшить, добавив фермент ДНК-лигазу, в результате чего получится кольцевая, ковалентно замкнутая молекула. У всех молекул ДНК, расщепленных одной эндонуклеазой, имеются одинаковые концы из 4–6 нуклеотидов, а сайт узнавания состоит из шести нуклеотидных пар. Мерц и Дэвис сделали вывод, что «...последовательно используя два фермента – рестрикгазу RI и ДНК-лигазу, – можно «перекомбинировать» любые две молекулы ДНК с RI-сайтами и получить гибридную молекулу». Это открытие стало ключевым в развитии технологии рекомбинантных ДНК, поскольку, по словам этих же ученых, указало «простой путь ...создания специфических рекомбинантных молекул ДНК in vitro».
При отборе трансформированных клеток руководствуются следующими соображениями. Если клетки, содержащие немодифицированную плазмиду pUC19, выращивать в присутствии изопропил-β-D-тиогалактопиранозида (ИПТГ), который является индуктором lас-оперона, то продукт гена lacl не сможет связаться с промоторно-операторной областью гена lacZ', и как следствие будут происходить транскрипция и трансляция плазмидного фрагмента гена lacZ'. Продукт этого фрагмента свяжется с белком, кодируемым хромосомной ДНК, и в результате образуется активная β-галакгозидаза. Последовательность с множеством сайтов рестрикции (полилинкер) встроена в ген lacZ' так, что она не влияет на продукцию функциональной Р-галактозидазы, и если в среде присутствует ее субстрат 5-бром-4-хлор-З-индолил-β-D-галактопиранозид (X-Gal), то он будет гидролизоваться под действием этого фермента с образованием продукта синего цвета, окрашивающего колонии клеток, содержащих немодифицированную плазмиду pUC19.
Для клонирования в pUC19 донорную ДНК расщепляют одной из рестриктаз, чей сайт находится в полилинкере; плазмидную ДНК гидролизуют такой же рестриктазой, а затем обрабатывают щелочной фосфатазой. Обе ДНК смешивают в присутствии ДНК-лигазы Т4 и используют образовавшийся продукт для трансформации клеток, которые могут синтезировать ту часть β-галактозидазы (LacZoc), которая соединяется с продуктом гена lacZ' с образованием активного фермента. Обработанные клетки высевают на питательную среду с ампициллином, ИПТГ и субстратом для β-галактозидазы. Нетрансфомированные клетки не могут расти в присутствии ампициллина, а клетки, несущие интактную плазмиду, образуют на среде с ампициллином колонии синего цвета. Клетки-хозяева, несущие гибридную плазмиду, образуют на той же самой среде белые колонии, поскольку обычно при встраивании в полилинкер чужеродной ДНК происходит прерывание кодирующей последовательности (сдвиг рамки считывания) гена lacZ', LacZ' не синтезируется и соответственно активная гибридная β-галактозидаза не образуется. В отсутствие активной β-галактозидазы клеточные колонии не превращают содержащийся в питательной среде субстрат в вещество синего цвета и остаются белыми. Далее проводят скрининг белых (положительных) колоний и последующую идентификацию тех из них, которые содержат искомую последовательность ДНК.
Помимо плазмид pBR322 и pUC, используется множество других векторов для клонирования, причем для некоторых из них существуют весьма остроумные системы отбора рекомбинантных клонов. Один из векторов на основе pUC содержит ген, кодирующий белок, который убивает клетку. При правильной рамке считывания этот «суицидальный» ген сшит с геном lacZ'. Клетка, несущая интактную плазмиду, на среде без ИПТГ не синтезирует «суицидальный» белок. Если в плазмиде присутствует вставка, а в питательную среду добавлен ИПТГ, то «суицидальный» белок продуцируется в неактивной форме, поскольку вставка, по всей вероятности, прерывает рамку считывания «сиуцидального» гена. Клетки с интактной плазмидой в присутствии ИПТГ синтезируют «суицидальный» белок и гибнут. Нетрансформированные клетки чувствительны к антибиотикам, в то время как рекомбинантные клоны несут ген, входящий в состав вектора, который обеспечивает их устойчивость к антибиотикам. Другими словами, в присутствии ИПТГ и антибиотика выживают только те клетки, которые несут плазмиду со вставкой. Один из «суицидальных» генов, который используется в таких системах отбора, кодирует фермент, препятствующий репарации двухцепочечных разрывов в хромосомной ДНК. Эти разрывы накапливаются, что влечет за собой гибель клеток.
Хотя некоторые векторы устроены весьма замысловато, все системы клонирования отвечают двум основным требованиям: наличие нескольких сайтов для клонирования и возможность достаточно простой идентификации клеток с рекомбинантными ДНК. Следует отметить, что уникальные сайты рестрикции выполняют в опытах с рекомбинантной ДНК двойную функцию. Они обеспечивают: 1) встраивание чужеродной ДНК в вектор; 2) вырезание клонированной последовательности из вектора. Другими словами, после встраивания фрагмента ДНК в определенный сайт этот фрагмент можно вырезать после клонирования той же самой рестриктазой, поскольку на концах встроенной нуклеотидной последовательности имеются два аналогичных сайта рестрикции. Иногда исходный сайт рестрикции модифицируется, тогда вырезание клонированного фрагмента затрудняется. Вырезанный фрагмент ДНК можно встраивать в векторы, предназначенные для определения нуклеотидной последовательности ДНК, или в векторы, специально сконструированные для обеспечения высокого уровня экспрессии (транскрипции и трансляции) клонированного гена.
Для всех рутинных процедур молекулярного клонирования широко используется Е. coli, но в качестве клеток-хозяев часто выступают и другие бактерии, например Bacillus subtilis и Agrobacterium tumefaciens. Во многих случаях в векторы, которые функционируют в Е. coli, можно встроить второй сайт инициации репликации, обеспечивающий их репликацию в других клетках. Эти так называемые челночные векторы позволяют сначала проводить клонирование в Е. coli, а затем внедрять готовую конструкцию в другие клетки. Кроме того, создано множество плазмидных векторов, которые содержат один сайт начала репликации ДНК для широкого спектра хозяев. Эти векторы можно использовать для работы с самыми разными микроорганизмами.
СОЗДАНИЕ И СКРИНИНГ БИБЛИОТЕК
Создание геномной библиотеки
Цель биотехнологических экспериментов часто состоит в идентификации генов, кодирующих определенные белки (структурных генов). У прокариот кодирующие домены структурных генов непрерывны, а у эукариот кодирующие области (экзоны) разделены некодирующими (нитронами). Соответственно при клонировании генов про- и эукариот должны применяться разные стратегии.
У прокариот искомая последовательность (ДНК-мишень) часто составляет минимальную часть (0,02%) суммарной хромосомной ДНК. Как же проводить клонирование и отбор столь редкого фрагмента ДНК? Чтобы решить эту проблему, суммарную ДНК гидролизуют рестриктазой и каждый из получившихся фрагментов встраивают в вектор. Теперь необходимо обнаружить специфическую клеточную линию (клон), которая несет нужную нам последовательность, выделить ее и охарактеризовать. Процесс разделения геномной ДНК на клонируемые элементы и введение этих элементов в клетки-хозяева называется созданием геномной библиотеки (банка клонов, банка генов). Полная библиотека по определению содержит весь геном данного организма.
Один из способов создания библиотеки ДНК состоит в обработке донорной ДНК рестрикгазой, узнающей тетрануклеотиды. Такой рестрикгазой является Sau3Al, которая вносит один разрыв примерно на 256 пар оснований. Гидролиз проводят в таких условиях, чтобы происходило лишь частичное расщепление, так что образуются фрагменты всевозможных размеров (рис. 4.10). Частичный гидролиз позволяет клонировать целые гены, однако, поскольку сайты рестрикции расположены не случайным образом, некоторые фрагменты могут оказаться слишком крупными для клонирования. В результате в распоряжении исследователя оказывается неполная библиотека, что может затруднить или даже сделать невозможным обнаружение искомой последовательности ДНК. Эту проблему можно решить, используя другую рестриктазу.
Следующий после создания библиотеки этап – это поиск клона (клонов), несущего искомую последовательность ДНК. Для этого используют три широко известных метода: гибридизацию с меченым ДНК-зондом с последующим радиоавтографическим анализом, иммунологический скрининг и скрининг по активности белка, кодируемого геном-мишенью.
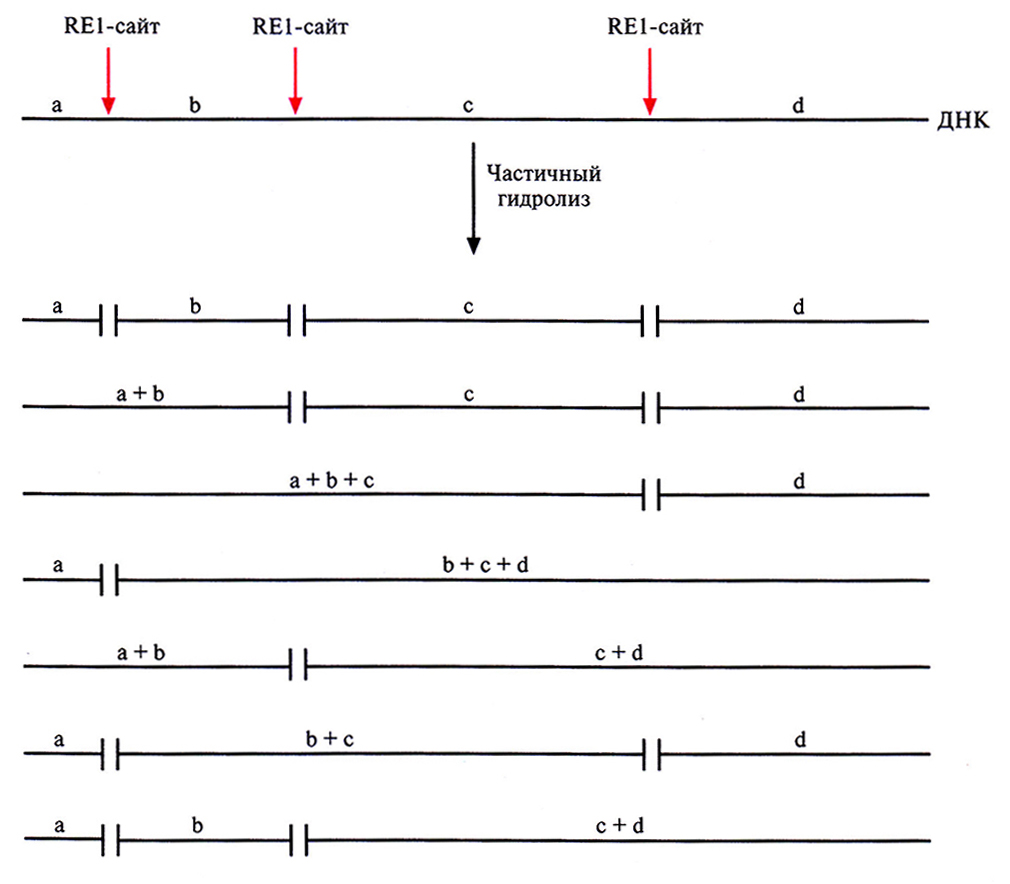
Рис. 4.10. Частичный гидролиз фрагмента ДНК рестрицирующей эндонуклеазой типа II. Глубину гидролиза обычно контролируют изменением времени инкубации или количества фермента. Одни молекулы, представленные на рисунке, расщеплены по всем сайтам для рестрицирующей эндонуклеазы 1 (RE1-сайтам), другие только по некоторым.
Скрининг с помощью гибридизации
Нужную нуклеотидную последовательность в образце ДНК можно обнаружить с помощью ДНК-зонда, спаривающегося только с искомой последовательностью. Для этого ДНК сначала переводят в одноцепочечную форму, подвергнув ее тепловой обработке или воздействию щелочью. В этих условиях водородные связи между основания ни разрываются и цепи расходятся (происходит денатурация). Если теперь медленно снизить температуру, то произойдет их воссоединение (ренатурация). При этом, если в растворе присутствует одноцепочечный ДНК-зонд, он тоже будет ренатурировать с ДНК, специфически спариваясь с комплементарными участками. В результате образуется гибридная ДНК, т. е. двухцепочечная молекула, цепи которой принадлежат двум разным ДНК.
Процедура ДНК-гибридизации состоит в следующем. ДНК-мишень подвергают денатурации и одноцепочечные молекулы необратимо «пришивают» к твердой подложке (нитроцеллюлозному или найлоновому фильтру). Эту процедуру обычно проводят при высокой температуре. Затем фильтр инкубируют с одноцепочечным ДНК-зондом, меченным радиоизотопом или другой меткой. Если нуклеотидные последовательности зонда и ДНК-мишени комплементарны, то происходит их спаривание (т. е. гибридизация) (рис. 4.11). Гибридные молекулы можно визуализировать радиоавтографическим (дополнение 4.2) или другим методом, зависящим от природы метки. Если комплементарность между зондом и ДНК-мишенью отсутствует, то гибридизации не происходит, и мы получаем отрицательный результат. Обычно размер зонда варьирует от 100 до 1000 п. н. и более, хотя можно использовать как более крупные зонды, так и зонды меньшего размера. Для гибридизации, т. е. для образования стабильного комплекса, необходимо, чтобы на участке длиной 50 нуклеотидов совпадало более 80% из них, но это зависит от условий реакции.
ДОПОЛНЕНИЕ 4.2
Радиоавтография
Этот метод широко применяется для локализации радиоактивного материала в клетке, срезе ткани или на пластине геля после электрофореза смеси макромолекул. Для регистрации радиоактивных зон на исследуемый образец накладывают рентгеновскую пленку, в которой под действием радиоактивного излучения из бромида серебра образуется металлическое серебро. «Засвеченные» участки, соответствующие радиоактивным зонам, наблюдаются визуально после проявления пленки. Одним из вариантов радиоавтографии является флюорография. В этом случае в исследуемый образец импрегнируют сцинтиллятор и вновь накладывают рентгеновскую пленку. Метод основан на том, что низкоэнергетические Р-частицы, образующиеся при распаде изотопа (например, трития), взаимодействуют с молекулами сцинтиллятора, при этом энергия радиоактивного распада преобразуется в световую энергию, которая и регистрируется рентгеновской пленкой, чувствительной к синей области спектра. Все операции при радиоавтографии необходимо проводить в темноте, чтобы не засветить пленку.
Очень важной областью применения радиоавтографии является обнаружение радиоактивного ДНК-зонда после его гибридизации с препаратом ДНК, подвергнутым электрофоретическому разделению. К сожалению, провести гибридизацию в самом геле невозможно, поскольку зонд не может в него проникнуть. Поэтому ДНК после электрофореза переносят на нитроцеллюлозный или найлоновый фильтр по методу Саузерна (Саузерн-блоттинг) или с помощью элюции. Расположение молекул ДНК на фильтре в точности соответствует таковому в геле. Перенесенную на фильтр ДНК подвергают денатурации и фиксируют, а затем проводят гибридизацию с радиоактивным ДНК-зондом. Гибридизационный сигнал регистрируют радиоавтографическими методами.
Перенос ДНК из геля на фильтр носит название Саузерн-блоттинга в честь Эдвина Саузерна, который изобрел этот метод. Использующиеся в литературе термины «Нозерн-блоттинг» и «Вестерн-блоттинг» относятся к переносу РНК и белков соответственно. Эти названия – в переводе «северный» и «западный» – не имеют никакого отношения к сторонам света и были придуманы остроумными коллегами Саузерна (Southern – южный). Тем самым они как бы напутствовали Саузерна на разработку новых методов переноса макромолекул, а также четко обозначили, о переносе каких макромолекул идет речь. Кроме того, все увидели, что шутить умеют не только физики, но и биологи.
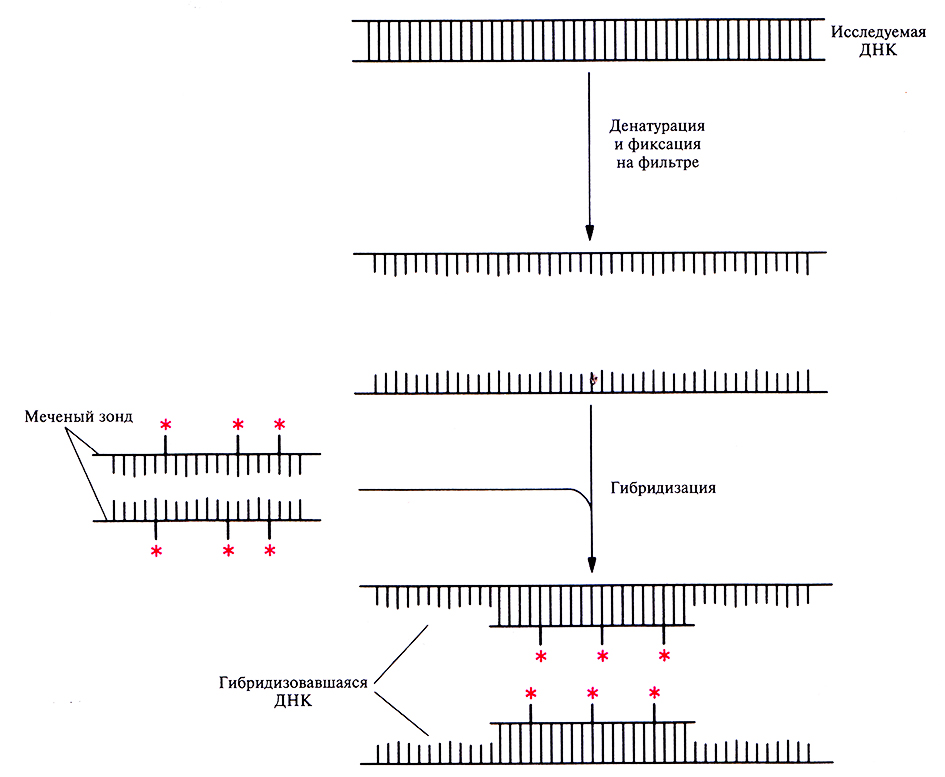
Рис. 4.11. ДНК-гибридизация. Исследуемую ДНК подвергают денатурации и фиксируют на твердой подложке, например на нитроцеллюлозном или найлоновом фильтре. Меченый ДНК-зонд (обычно длиной от 100 до 1000 п. н.) тоже денатурируют, наносят на фильтр с исследуемой ДНК и проводят их отжиг. Для удаления негибридизовавшегося ДНК-зонда фильтр промывают и визуализируют метку. Если гибридизация между зондом и исследуемой ДНК не произошла, то никакой метки на фильтре не обнаруживается. (Метка на рисунке обозначена цветной звездочкой.)
Меченые ДНК-зонды можно получить разными способами. Один из них, называемый методом случайных праймеров, основан на применении смеси синтетических олигонуклеотидов (олигомеров), содержащих все возможные комбинации из шести нуклеотидов. Некоторые из этих олигонуклеотидов оказываются комплементарными последовательностям ДНК-мишени и гибридизуются с ними, если ДНК предварительно денатурировать (рис. 4.12). После отжига олигонуклеотидов с денатурированной ДНК-матрицей в реакционную смесь добавляют четыре дезоксирибонуклеотида (дезоксирибонуклеозидтрифосфаты; dNTP), один из них – меченый, и фрагмент ДНК-полимеразы 1 Е. coli (фрагмент Кленова). Фрагмент Кленова обладает ДНК-полимеразной и З'-экзонуклеазной активностями, но не 5'-экзонуклеазной активностью, присущей ДНК-полимеразе I Е. coli, которая могла бы расщепить новосинтезированные молекулы ДНК. Одиночные цепи ДНК-мишени служат матрицами для синтеза новых молекул ДНК, а связанные с ними случайным образом олигонуклеотиды – затравками (рис. 4.12). При радиоактивном мечении один из dNTP содержит α-32Р, так что 32Р-меченным оказывается и сам зонд. Радиоактивную метку выявляют с помощью радиоавтографии.
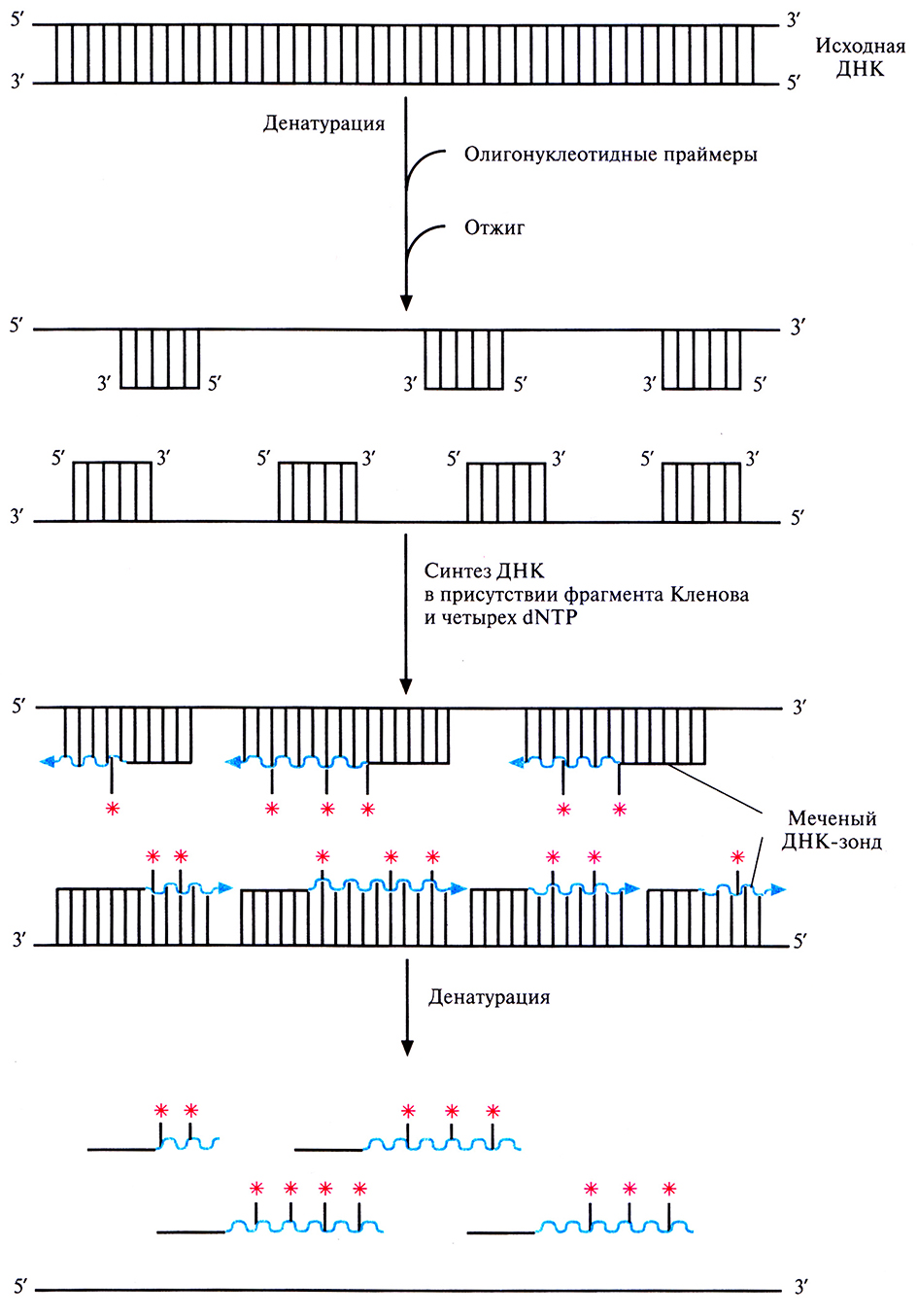
Рис. 4.12. Получение меченого ДНК-зонда методом случайных праймеров. К денатурированной двухцепочечной ДНК, содержащей нуклеотидную последовательность, которую предполагается использовать в качестве зонда, добавляют гексануклеотиды (смесь всех возможных комбинаций из шести нуклеотидов) и отжигают смесь. Некоторые из олигонуклеотидов гибридизуются с немеченой денатурированной ДНК, и в присутствии фрагмента Кленова и четырех dNTP (один из которых меченый [*]) служат затравкой для синтеза комплементарной цепи. После денатурации синтезированной ДНК получают смесь меченых фрагментов ДНК, которые вместе составляют практически полноразмерную исходную ДНК-матрицу.
В качестве нерадиоизотопной метки часто используют биотин, который присоединяют к одному из четырех dNTP. Для выявления гибридизовавшегося биотинилированного зонда на фильтр наносят конъюгат стрептавидина с соответствующим ферментом (например, щелочной фосфатазой). Стрептавидин образует комплекс с биотином, который обнаруживается благодаря тому, что под действием фермента образуется окрашенное или люминесцирующее вещество – продукт превращения нанесенного на фильтр субстрата.
Зонды для скрининга геномной библиотеки можно получить по крайней мере двумя способами. Во-первых, можно использовать клонированную ДНК близкородственного организма (гетерологичный зонд). В этом случае условия гибридизации нужно подбирать таким образом, чтобы она могла происходить при существенном расхождении между нуклеотидными последовательностями зонда и искомой ДНК; это позволяет решить проблемы, связанные с заведомым различием между ДНК – источником зонда и исследуемой ДНК. Во-вторых, зонд можно получить методом химического синтеза, основываясь на известной аминокислотной последовательности белкового продукта искомого гена.
Скрининг библиотек геномных ДНК обычно проводят по следующей схеме. После трансформации высевают клетки на чашки с питательной средой и переносят выросшие колонии на твердую подложку (например, на нитроцеллюлозный или найлоновый фильтр); проводят лизис клеток, затем депротеинизацию и денатурацию ДН К; фиксируют ДНК на подложке. Наносят на фильтр меченый зонд и проводят отжиг, а затем радиоавтографию. Колонии на исходной чашке, которые содержат гибридизовавшуюся ДНК, выделяют и культивируют (рис. 4.13). Поскольку большинство библиотек получается в результате частичного гидролиза, положительный гибридизационный сигнал может быть получен для нескольких колоний (клонов). Теперь необходимо определить, какой именно клон (если таковой имеется) содержит искомый ген целиком. С помощью гель-электрофореза и картирования определяют размер каждого фрагмента (вставки) и идентифицируют аналогичные фрагменты или фрагменты с перекрывающимися последовательностями. Можно также провести дополнительное клонирование с тем, чтобы составить полный ген из перекрывающихся фрагментов. Или, если вставка в каком-либо из клонов достаточно велика и вполне может содержать весь ген, провести ее секвенирование и убедиться в наличии старт- и стоп-кодонов и полноразмерной нуклеотидной последовательности, кодирующей искомый белок.
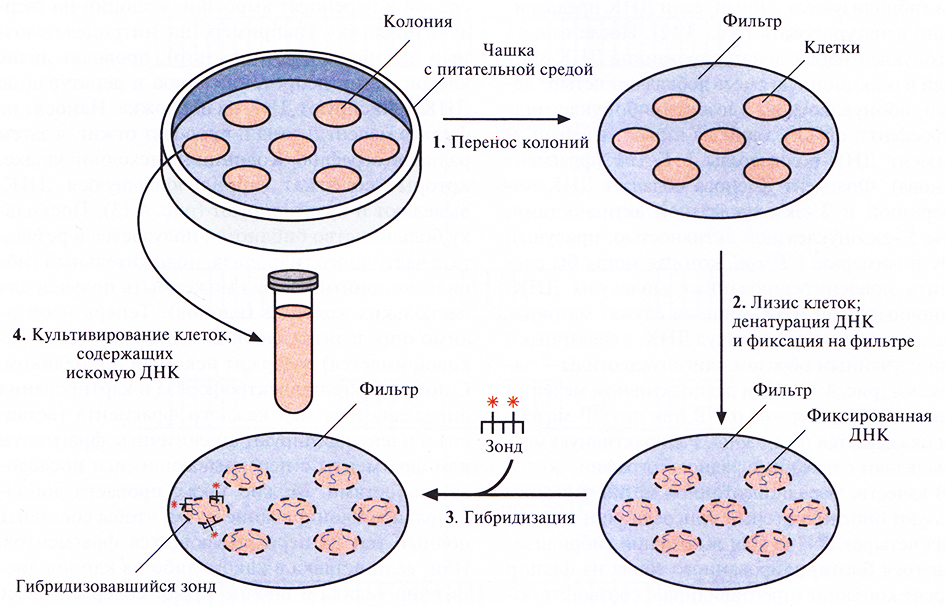
Рис. 4.13. Скрининг библиотеки геномной ДНК с применением меченого зонда. Клетки после трансформации высевают на твердую питательную среду, обеспечивающую рост только трансформированные клеток. 1. Переносят клетки из каждой выросшей колонии на твердую подложку (например, нитропеллюлозный или найлоновый фильр) так, чтобы их расположение соответствовало таковому на чашке. 2. Клетки лизируют, высвободившуюся ДНК подвергают денатурации и депротеинизации и фиксируют на фильтре. 3. На фильтр наносят меченый ДНК-зонд и проводят гибридизацию. Смывают с фильтра негибридизовавшийся зонд и проводят радиоавтографию с тем чтобы определить, какие клетки содержат меченый ДНК-зонд. 4. Идентифицируют на чашке колонии, содержащие искомую ДНК (положительный гибридизационный сигнал), отбирают из них материал и культивируют.
К сожалению, никто не может дать гарантии, что в библиотеке представлена вся нуклеотидная последовательность нужного гена. Если поиск полноразмерного гена оказался безрезультатным, можно создать другую библиотеку, используя другую рестриктазу, и провести скрининг с помощью исходного зонда или зондов, созданных на основе предыдущей библиотеки. Чтобы повысить вероятность присутствия в библиотеке полной версии искомого гена, можно также создать библиотеки, содержащие фрагменты ДНК заведомо большего размера, чем средний размер прокариотического гена (этот вариант мы рассмотрим в данной главе позже).
Иммунологический скрининг
В отсутствие ДНК-зонда для скрининга геномной библиотеки можно использовать другие методы. Например, если клонированный ген экспрессируется, то его продукт – весь белок или его часть – можно обнаружить иммунологическими методами. Технически эта процедура имеет много общего с гибридизацией. Все клеточные линии (клоны) библиотеки высевают на чашки с питательной средой. Выросшие колонии переносят на фильтр, клетки лизируют, а высвободившиеся белки фиксируют на фильтре. Затем на фильтр наносят первые антитела, которые специфически связываются с данным белком (антигеном), все несвязавшиеся антитела удаляют, а фильтр помещают в раствор вторых антител, специфичных в отношении первых антител. Во многих тест-системах используют конъюгаты вторых антител с ферментом, например с щелочной фосфатазой. После отмывания фильтра добавляют бесцветный субстрат. Если вторые антитела связываются с первыми, то под действием фермента происходит гидролиз субстрата с образованием окрашенного вещества в том месте, где идет реакция (рис. 4.14).
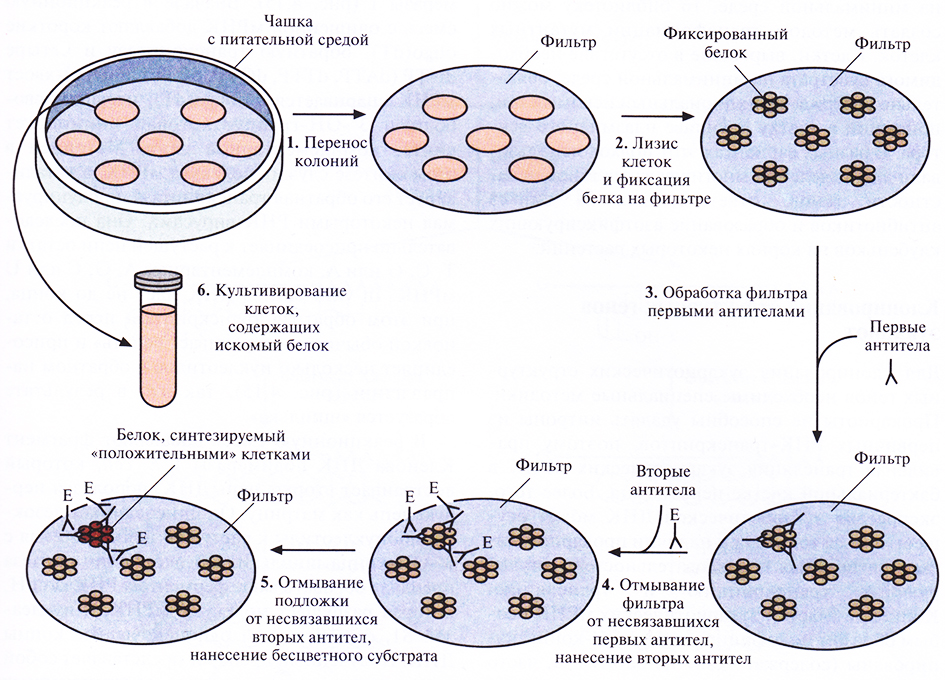
Рис. 4.14. Иммунологический скрининг геномной библиотеки (иммунологическое тестирование колоний). Клетки после транстформации высевают на твердую питательную среду, обеспечивающую рост только трансформированных клеток. 1. Переносят клетки из каждой выросшей колонии на твердую подложку (например, нитроцеллюлозныи или наилоновый фильтр) так, чтобы их расположение в точности соответствовало таковому на чашке. 2. Клетки подвергают лизису, белки фиксируют на фильтре. 3. Наносят на фильтр первые антитела, которые связываются только с искомым белком. 4. Несвязавшиеся первые антитела удаляют, наносят на фильтр вторые антитела, специфичные в отношении первых антител, связанные с ферментом (например, щелочной фосфатазой). 5. Смывают с фильтра все несвязавшиеся вторые антитела и наносят на него бесцветный субстрат, при гидролизе которого образуется окрашенный продукт. Гидролиз может произойти только в присутствии вторых антител. 6. Отбирают колонии на чашке, соответствующие окрашенным пятнам на фильтре, и культивируют их. В них может содержаться рекомбинантная ДНК, кодирующая белок, гомологичный первым антителам.
Скрининг по активности белка
Метод гибридизации ДНК и иммунологические методы позволяют идентифицировать многие гены и их продукты. Если при этом искомый ген кодирует фермент, не синтезируемый клеткой-хозяином, то для обнаружения клонов, содержащих данный ген, можно использовать метод идентификации на чашках. Так были идентифицированы гены α-амилазы, эндоглюканазы и β-галактозидазы различных организмов. Для этого клоны Е. coli, составляющие геномную библиотеку данных организмов, высевали на чашках с питательной средой, содержащей специфический субстрат. После окрашивания селективным красителем клетки, способные утилизировать этот субстрат, приобретали характерную окраску.
Если искомый ген кодирует продукт, без которого мутантная клетка-хозяин не может расти на минимальной среде, то библиотеку можно создать методом трансформации мутантных клеток. Клетки, выросшие в отсутствие необходимого субстрата на минимальной среде, обязательно содержат функциональный искомый ген, попавший в клетку в составе плазмидного вектора. В разных вариантах этот подход использовали для выделения многих важных генов, в частности генов, ответственных за синтез антибиотиков и образование азотфиксирующих клубеньков на корнях некоторых растений.
КЛОНИРОВАНИЕ СТРУКТУРНЫХ ГЕНОВ ЭУКАРИОТ
Для клонирования эукариотических структурных генов необходимы специальные методики. Прокариоты не способны удалять интроны из первичных РНК-транскриптов, поэтому правильная трансляция эукариотических мРНК в бактериальной клетке невозможна. Более того, экспрессия эукариотической ДНК может осуществляться только при наличии прокариотических сигнальных последовательностей, регулирующих транскрипцию и трансляцию. Концевые участки эукариотических мРНК особым образом модифицированы: их 5'-концы кэпированы (содержат «кэп» из остатка G, часто метилированного), а 3'-концы полиаденилированы (содержат ро1у(А)-«хвост» из примерно 200 остатков аденозина).
Наличие poly (А)-хвоста позволяет отделить мРНК от рибосомной и транспортной РНК. Для этого суммарную эукариотическую РН К пропускают через колонку, заполненную целлюлозой, к которой «пришиты» короткие олигонуклеотидные цепочки из тимидиновых остатков длиной примерно 15 звеньев, oligo(dT). Poly(A)-хвосты молекул мРНК спариваются с oligo(dT) и задерживаются в колонке, а молекулы тРНК и рРНК свободно проходят через нее. Затем колонку промывают буфером, в котором происходит разрыв водородных связей между А и Т, и мРНК высвобождается.
Саму мРНК нельзя встроить в ДНК-вектор, сначала на ней необходимо синтезировать двухцепочечную ДНК. Для этого последовательно используют две разные полимеразы: обратную транскриптазу и фрагмент Кленова ДНК-поли меразы I (рис. 4.15). Вначале в реакционную смесь с очищенной мРНК добавляют короткие oligo(dT), обратную транскриптазу и четыре dNTP (dATP, dTTP, dGTP, dCTP). Ро1у(А)-хвост мРНК спаривается с oligo(dT), несушим свободную 3'-ОН-группу, которая инициирует синтез комплементарной цепи. Матрицей в этом синтезе служит молекула мРНК, а катализирует его обратная транскриптаза, продуцируемая некоторыми РНК-вирусами. Она последовательно присоединяет к растущей цепи остатки Т, С, G или А, комплементарные A, G, С или U мРНК. In vitro синтез ДНК идет не до конца, при этом обратная транскриптаза перед остановкой обычно «поворачивает вспять» и присоединяет несколько нуклеотидов в обратном направлении (рис. 4.15), так что в результате образуется «шпилька».
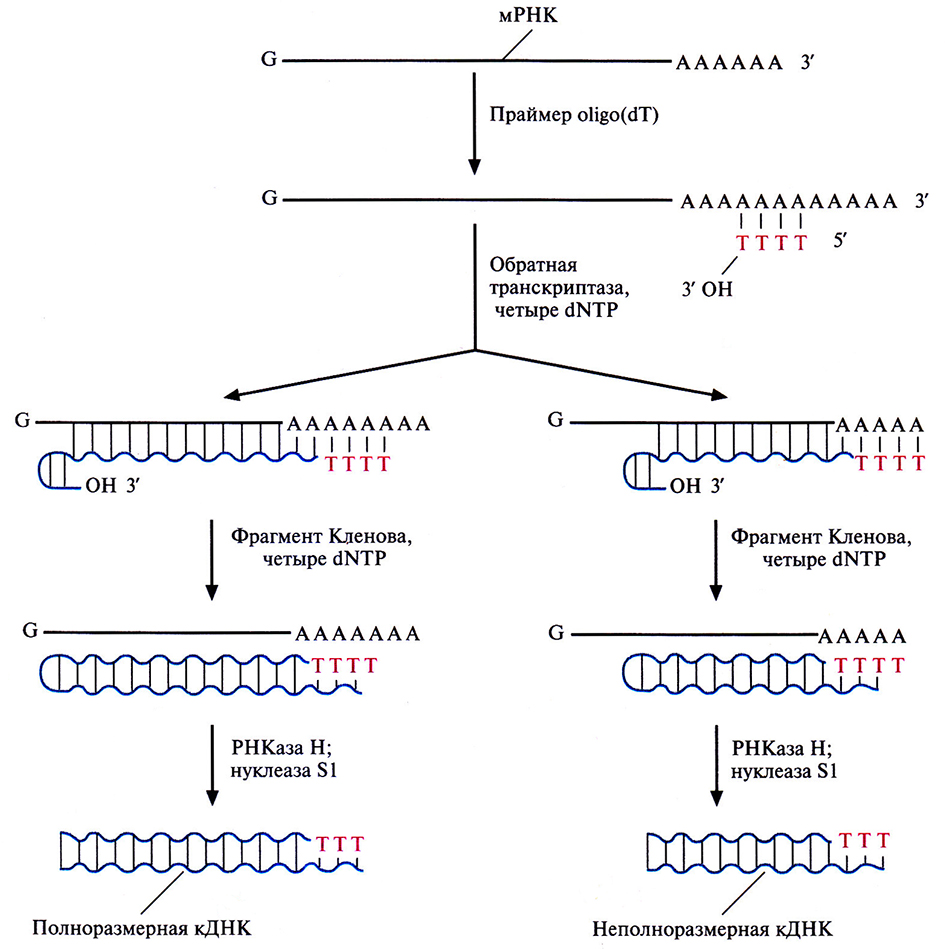
Рис. 4.15. Синтез кДНК. К препарату очищенной мРНК добавляют праймер oligo(dT). Для синтеза ДНК на РНК-матрице используют фермент обратную транскриптазу и четыре dNTP. In vitro обратная транскриптаза не обеспечивает синтез полноразмерных кДНК-копий на всех матрицах и образует на конце растущей цепи шпильку со свободной З'-ОН-группой. Эта группа инициирует синтез второй цепи ДНК при участии фрагмента Кленова. После завершения синтеза молекулы мРНК гидролизуют РНКазой Н, а ДНК обрабатывают нуклеазой S1, в результате чего получаются линейные молекулы ДНК с тупыми концами без шпилек.
В реакционную смесь добавляют фрагмент Кленова ДНК-полимеразы I Е. coli, который достраивает вторую цепь ДНК, используя первую цепь как матрицу. Он присоединяет дезоксирибонуклеотиды к растущей цепи, начиная с З'-ОН-конца шпильки. По окончании синтеза препарат обрабатывают ферментом РНКазой Н, которая разрушает молекулы мРНК, и нуклеазой S1, отщепляющей одноцепочечные концы ДНК. Полученный препарат представляет собой смесь частично и полностью двухцепочечных комплементарных ДНК-копий (кДНК) мРНК, преобладающей в исходном образце.
Разные кДНК можно встроить в плазмидный вектор и получить кДНК-библиотеку. Для скрининга кДНК-библиотеки с целью идентификации клонов, несущих специфические гибридные плазмиды, можно использовать метод гибридизации или иммунологические методы. В последнем случае кДН К должна быть встроена в сайт, находящийся под контролем бактериального промотора, обеспечивающего транскрипцию. Однако практически ни один вектор не гарантирует, что во встроенной кДНК сохранится правильная рамка считывания и синтезируется правильная полипептидная цепь. Тем не менее все положительные клоны, выявленные тем или иным методом, необходимо подвергнуть дальнейшей проверке и идентифицировать те из них, которые несут полноразмерную нуклеотидную последовательность, кодирующую белок-мишень.
ВЕКТОРЫ ДЛЯ КЛОНИРОВАНИЯ КРУПНЫХ ФРАГМЕНТОВ ДНК
Векторы на основе бактериофага λ
С помощью плазмидных векторов можно клонировать фрагменты ДНК длиной до 10 т. п. н. Однако при создании геномных библиотек часто приходится работать с более крупными фрагментами. Для этого были разработаны векторы на основе бактериофага А. Е. coli.
После проникновения фага к в клетку Е. coli события могут развиваться по двум сценариям. Если реализуется литический цикл, то фаг начинает интенсивно размножаться и примерно через 20 мин клетка разрушается (лизирует) с высвобождением до 100 новых фаговых частиц. При альтернативном варианте развития событий фаговая ДНК включается в хромосому Е. coli как профаг и реплицируется в клетке вместе с нормальными бактериальными генами (состояние лизогении). Однако при недостатке питательных веществ или иных неблагоприятных обстоятельствах интегрированная фаговая ДНК высвобождается, и запускается литический цикл развития. Размер ДНК фага X составляет примерно 50 т. п. н., причем значительная ее часть (около 20 т. п. н.) несущественна для размножения фага и отвечает за его встраивание в хозяйскую ДНК. В связи с этим возникла идея, что ее можно заменить фрагментом другой ДНК эквивалентного размера. Образующаяся рекомбинантная молекула будет реплицироваться в клетке как ДНК «рекомбинантного» фага X, «вставшего» на литический путь развития.
Чтобы понять, как функционирует векторная система на основе фага X, необходимо рассмотреть молекулярные аспекты литического цикла развития. Инфекционная фаговая частица имеет головку, в которой заключена плотно упакованная ДНК длиной примерно 50 т. п. н., и отросток с отходящими от него тонкими белковыми нитями (фибриллами). Сборка головки и отростка и упаковка ДНК четко скоординированы. ДНК фага X – это линейная двухцепочечная молекула длиной 50 т. п. н. с одноцепочечными 5'-«хвостами» из 12 нуклеотидов. Их называют липкими (cos) концами, поскольку они взаимно комплементарны и могут спариваться друг с другом. После того как фаговая ДНК проходит через отросток и попадает в Е. coli, cos-концы соединяются с образованием кольцевой молекулы. На раннем этапе литического цикла в результате репликации кольцевой молекулы ДНК образуется линейная молекула, состоящая из нескольких сегментов длиной 50 т. п. н. (рис. 4.16,А). Каждый из таких сегментов упаковывается в белковую головку, к последней присоединяется уже собранный отросток и образуется новая фаговая частица (рис. 4.16,Б). При упаковке молекулы ДНК длиной менее 38 т. п. н. получается неинфекционная фаговая частица, а фрагменты длиной более 52 т. п. н. не умещаются в головку. Сегменты длиной 50 т. п. н. в линейной молекуле ДНК разделены cos-сайтами, и именно по этим сайтам разрезается молекула, когда очередной сегмент заполняет головку. Разрезание осуществляет фермент, находящийся у входа в головку.
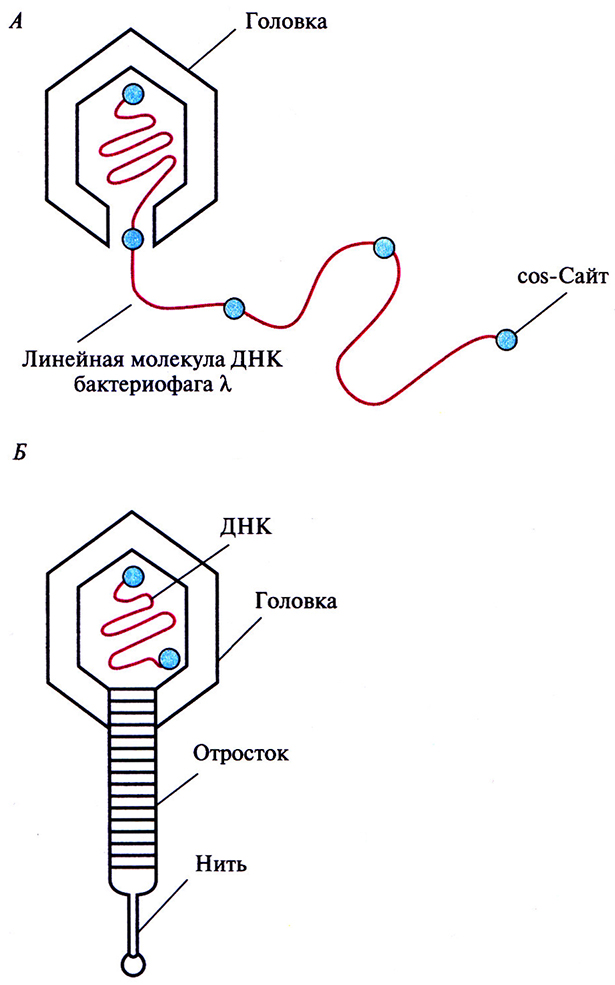
Рис. 4.16. Литический путь развития бактериофага X. А. При репликации кольцевой ДНК бактериофа1 а X образуется линейная молекула, состоящая из повторяющихся сегментов длиной примерно 50 т. п. н. Каждый из этих сегментов представляет собой полноразмерную фаговую ДНК. Б. Фаговая головка вмещает один такой сегмент, затем к головке присоединяется уже собранный отросток.
В результате исследований по изучению сборки фага X была разработана система упаковки молекул ДНК in vitro с образованием инфекционных фаговых частиц. Смешав в пробирке очищенные пустые головки, фаговую ДНК и собранные отростки, можно получить инфекционные фаговые частицы.
Один из множества λ-векторов для клонирования имеет два ВатHll-сайта, фланкирующих участок длиной 20 т. п. н. При гидролизе очищенной фаговой ДНК рестриктазой ВатHll образуется три фрагмента. Так называемое левое плечо (область L) содержит генетическую информацию о головке и отростке фага, правое плечо (область R) ведает репликацией ДНК и лизисом, а средний сегмент имеет гены, ответственные за процессы интеграции и исключения (сегмент l/Е, от англ. integration/excision). Задача исследователя состоит в том, чтобы заменить этот центральный участок нуклеотидной последовательностью длиной примерно 20 т. п. н. (рис. 4.17) нужной ДНК. ДНК, предназначенную для клонирования, тоже расщепляют с помощью ВатHll и выделяют фрагменты размером от 15 до 20 т. п. н. Оба препарата – фаговую и чужеродную ДНК – объединяют и добавляют ДНК-лигазу фага Т4, а затем – пустые головки и уже собранные отростки. Фрагменты ДНК длиной 50 т. п. н. упаковываются в головки, к ним присоединяются отростки и образуются инфекционные фаговые частицы. Фрагменты большего (>52 т. п. н.) или меньшего (<38 т. п. н.) размера упаковываться не могут. Рекомбинантный фаг X может размножаться только в тех штаммах Е. coli, которые не обеспечивают размножения фага λ с интактной областью 1/Е. Для сохранения рекомбинантного фага λ его периодически пересевают на свежую культуру Е. coli.
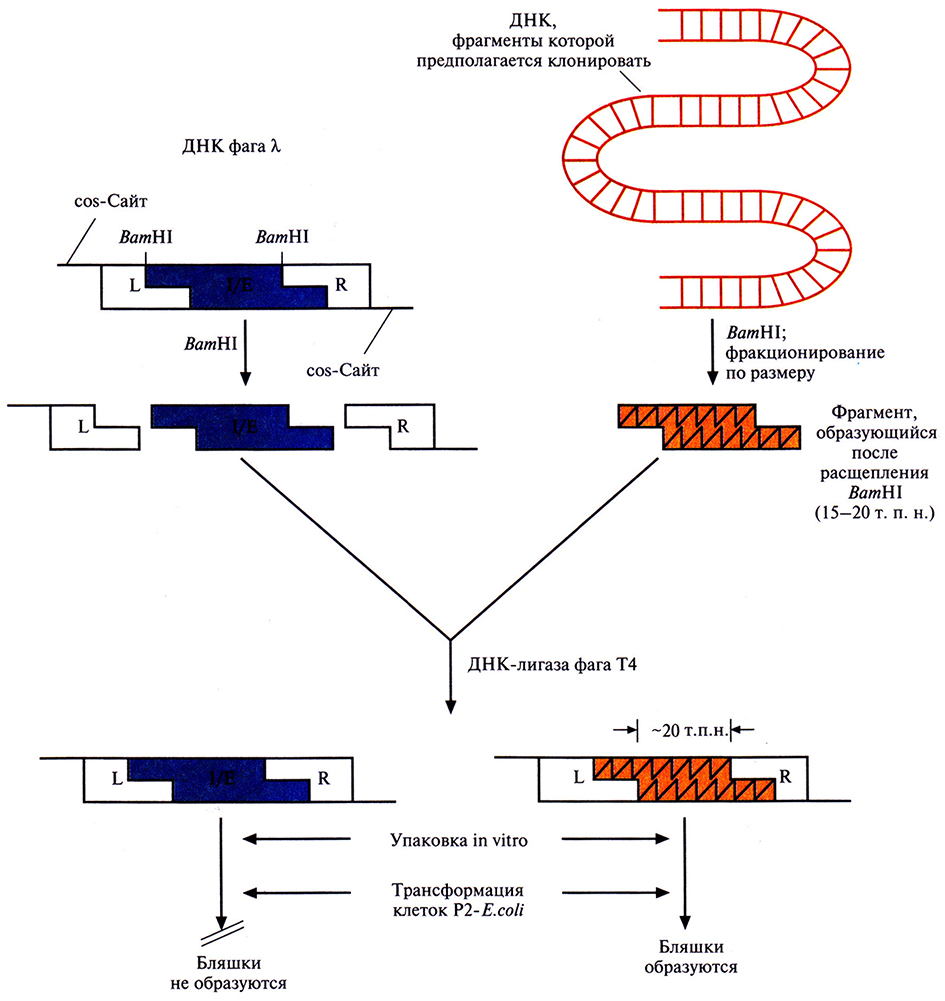
Рис. 4.17. Клонирующая система на основе бактериофага λ. Фаговая ДНК имеет два ВатHll-сайта, фланкирующих ее I/E-сегмент. Клонируемую ДНК расщепляют с помощью ВатHll, фракционируют полученные фрагменты по размеру и выделяют те из них, которые имеют размер от 15 до 20 т. п. н. Фаговую ДНК обрабатывают этим же ферментом. Оба препарата ДНК смешивают и обрабатывают ДНК-лигазой фага Т4. Лигированная смесь содержит самые разные комбинации ДНК, в том числе 1) восстановленную ДНК фага λ и 2) рекомбинантные молекулы, содержащие R- и L-области фаговой ДНК и вставку клонируемой ДНК размером -20 т. п. н., занявшую место области I/E фагового генома. Рекомбинантные молекулы упаковывают в головки бактериофага X in vitro, и после добавления отростков получают инфекционные фаговые частицы В инфицированных рекомбинантным фагом клетках Е. coli, в хромосому которых интегрирована ДНК бактериофага Р2, могут реплицлроваться и образовывать инфекционные частицы только молекулы ДНК, составленные из R- и L-областей фаговой ДНК и клонированной вставки размером ~20 т.п.н.
Для скрининга библиотек на основе фага X можно использовать ДНК-зонды или иммунологические методы. Зоны лизиса (бляшки) переносят на фильтр и соответствующим образом тестируют. Если используется ДНК-гибридизация, то вначале удаляют фаговые белки, затем ДНК денатурируют и фиксируют на фильтре. При тестировании иммунологическим методом белки, кодируемые клонированными генами, переносят и фиксируют на фильтре вместе с бляшкой. Сопоставив пятна на фильтре, дающие положительную реакцию, с бляшками на исходной чашке, отбирают позитивные бляшки и проводят субкультивирование. Субкультуры служат источником рекомбинантных бактериофагов, которые можно по отдельности культивировать в Е. coli.
Космиды
Векторы, называемые космидами, могут включать до 40 т. п. н. чужеродной ДНК и при этом активно амплифицироваться в Е. coli как плазмиды. Космиды объединяют в себе свойства плазмидных векторов и векторов на основе фага X. Например, широко применяемая космида pLFR-5 (приблизительно 6 т. п. н.) имеет два cos-еайта фага λ, разделенных сайтом рестрикции для Scal, полилинкер с шестью уникальными сайтами рестрикции (Hindlll, Pstl, Sail, BamHl, Smal и EcoRI), точку начала репликации ДНК (ori) и ген устойчивости к тетрациклину (Tetr). Эта космида может интегрировать чужеродную ДНК длиной до 40 т. п. н. (рис. 4.18). Предназначенные для клонирования фрагменты ДНК длиной около 40 т. п. н. очищают центрифугированием в градиенте плотности сахарозы от продуктов частичного гидролиза донорной ДНК рестриктазой BamHl (рис. 4.18), a pLFR-5 сначала подвергают гидролизу с помощью Seal, а затем BamHl. Препараты ДНК смешивают и лигируют. Те продукты лигирования, которые содержат вставку длиной 40 т. п. н., имеют суммарный размер, близкий к 50 т. п. н., и следовательно, могут упаковываться in vitro в головки фага λ. Реассоциировавшие молекулы pLFR-5, не содержащие вставок, упакованы не будут. После сборки фаговых частиц инфицируют ими Е. coli (рис. 4.18).
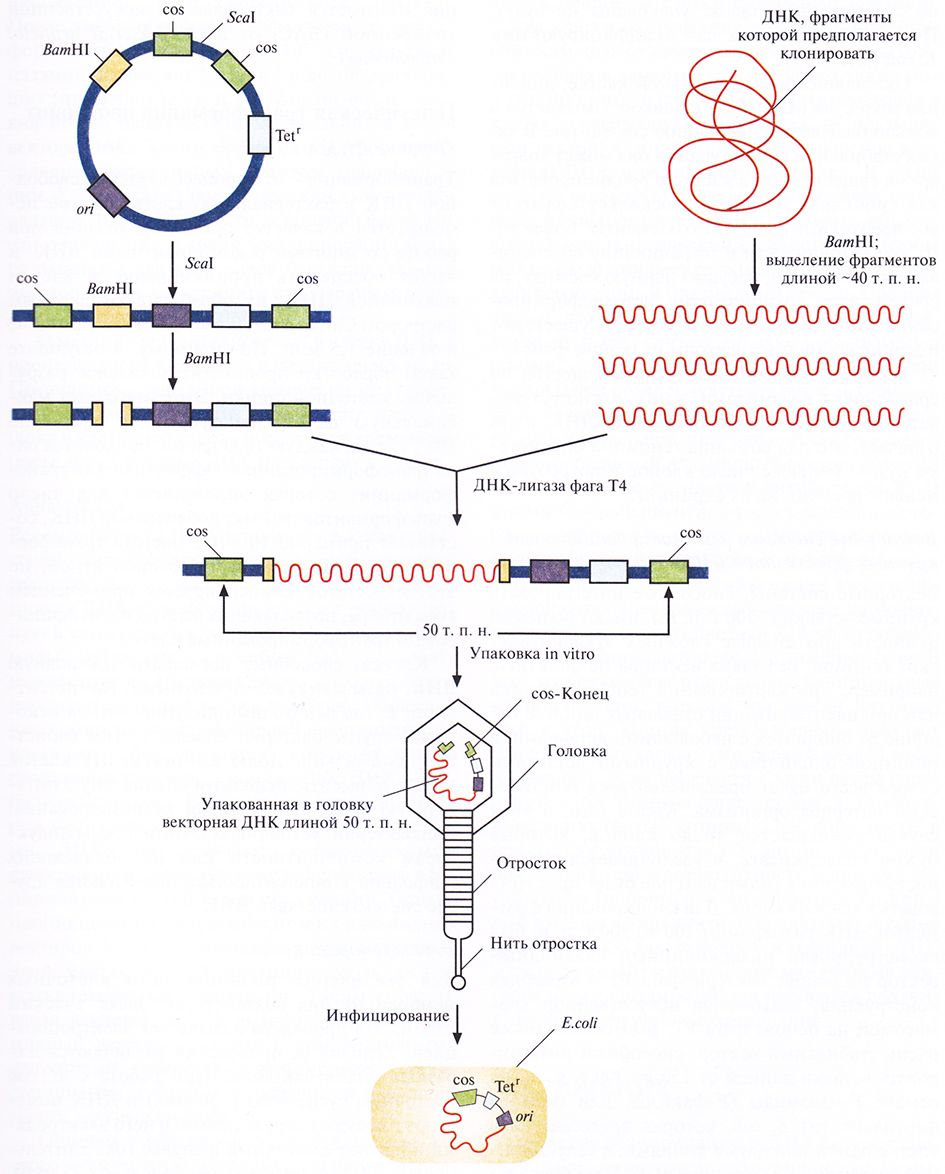
Рис. 4.18. Клонирование с помощью космидного вектора. Космида имеет точку начала репликации (ori), обеспечивающую ее существование в Е. coli в виде плазмиды; два интактных cos-конца, разделенных уникальным сайтом для Seal: ВатHI-сайт вблизи одного из cos-сайтов и ген устойчивости к тетрациклину (Tetr). ДНК, которую хотят клонировать, расщепляют рестриктазой BamHl и фракционируют по размеру, чтобы выделить молекулы длиной примерно 40 т. п. н. Плазмидную ДНК расщепляют с помощью Seal и Ват HI. Оба препарата ДНК смешивают и обрабатывают ДНК-лигазой фага Т4. Некоторые из гибридных молекул, образовавшихся после лигирования, содержат вставку размером около 40 т. п. н., так что их суммарная длина составляет примерно 50 т. п. и. Эти молекулы упаковываются in vitro в головки бактериофага X, затем к головкам прикрепляются отростки, и образуются инфекционные частицы. При инфицировании этим «фагом» Е. coli в бактериальной клетке оказывается линейная молекула ДНК с cos-концами, которые спариваются друг с другом ДНК-лигаза клетки-хозяина зашивает одноцепочечные разрывы, и образовавшаяся кольцевая молекула существует в клетке-хозяине как автономно реплицирующаяся единица. Трансформированные клетки можно идентифицировать по признаку устойчивости к тетрациклину.
Оказавшись в бактериальной клетке, линейная молекула pLFR-5 со вставкой замыкается в кольцо благодаря спариванию cos-сайтов. В такой стабильной конфигурации она может долгое время существовать в клетке и реплицироваться как гибридная плазмида, поскольку содержит все необходимые для этого элементы. Более того, ген устойчивости к тетрациклину обеспечивает рост колоний, несущих данную космиду, на среде с этим антибиотиком; нетрансформиро ванные клетки при этом погибают. Существуют и другие космидные векторы на основе фага λ.
Космиды имеют большое преимущество по сравнению с плазмидами: в них можно встраивать более протяженные фрагменты ДНК, а это означает, что для создания геномной библиотеки нужно меньшее число клонов и потребуется меньше времени на их скрининг.
Векторные системы для клонирования
очень крупных фрагментов ДНК
Векторные системы, способные интегрировать крупные вставки (>100 т. п. н.), имеют большую ценность при анализе сложных эукариотических геномов. Без таких векторов не обойтись, например, при картировании генома человека или при идентификации отдельных генов. В отличие от библиотек с небольшими вставками, в геномной библиотеке с крупными вставками скорее всего будет представлен весь генетический материал организма. Кроме того, в этом случае уменьшается число клонов, которые нужно поддерживать, и увеличивается вероятность того, что каждый из генов будет присутствовать в «своем» клоне. Для клонирования фрагментов ДНК размером от 100 до 300 т. п. н. был сконструирован низкокопийный плазмидный вектор на основе бактериофага Рl – химерная конструкция, называемая искусственной хромосомой на основе фага Рl. Был создан также очень стабильный вектор, способный интегрировать вставки длиной от 150 до 300 т. п. н., на основе F-плазмиды (F-фактора, или фактора фертильности) Е. coli, которая представлена в клетке одной или двумя копиями, с селекционной системой lacZ' векторов pUC. Эта конструкция называется бактериальной искусственной хромосомой (ВАС, от англ. bacterial artificial chromosomes).
ГЕНЕТИЧЕСКАЯ ТРАНСФОРМАЦИЯ ПРОКАРИОТ
Перенос ДНК в Е. coli
Трансформация – это процесс введения свободной ДНК в бактериальную клетку. Е. coli используется в качестве организма-хозяина при работе со многими рекомбинантными ДНК, и чтобы обеспечить проникновение в клетки плазмидной ДНК, их обрабатывают ледяным раствором СаС12, а затем выдерживают при 42 °С в течение 1,5 мин. По-видимому, в результате такой обработки происходит локальное разрушение клеточной стенки. Этот метод дает максимальную частоту трансформации, примерно 10–3, т. е. на каждую 1000 клеток приходится одна трансформированная. Эффективность трансформации, которая определяется как число грансформантов на 1 мкг добавленной ДНК, составляет примерно 107–108. Частота трансформации никогда не бывает 100-процентной, но этот недостаток компенсируется применением схем отбора, позволяющих быстро идентифицировать трансформированные клетки.
Клетки, способные поглощать чужеродную ДНК, называются компетентными. Компетентность Е. coli необходимо индуцировать, а некоторые другие бактерии обладают этим свойством изначально. Долю компетентных клеток можно повысить, используя специальную питательную среду или условия культивирования. Для бактерий, устойчивых к химическим индукторам компетентности или не обладающих природной компетентностью, применяются другие системы доставки ДНК.
Электропорация
Для увеличения проницаемости клеточных мембран на них воздействуют электрическим током. Эта процедура называется электропорацией. Условия ее проведения различаются для разных видов бактерий. При работе с Е. coli клеточную суспензию (~50 мкл) и ДНК помещают в сосуд с погруженными в него электродами и подают единичный импульс тока длительностью ~4,5 мс (емкость конденсатора 25 мкФ, напряжение 2,5 кВ, сопротивление 200 Ом). После такой обработки эффективность трансформации повышается до 109 для коротких плазмид (примерно 3 т. п. н.) и до 106 для больших (примерно 135 т. п. н.). Аналогичные условия можно использовать для введения в Е. coli вектора ВАС. Таким образом, электропорация является эффективным методом трансформации Е. coli плазмидами, содержащими вставки длиннее 100 т. п. н. Есть основания полагать, что подходящие условия электропорации будут разработаны для всех видов бактерий, так что эта процедура может стать стандартным методом трансформации.
О механизме проникновения в клетку ДНК в процессе электропорации известно очень мало. По-видимому, как и при химически индуцированной трансформации, в результате электрошока в клеточной стенке образуются временные поры, через которые ДНК и проходит в клетку.
Конъюгация
Рекомбинантная ДНК проникает в клетки бактерий, характеризующихся низкой частотой трансформации, таким же образом, как плазмидная ДНК из донорской клетки в реципиентную в естественных условиях. Некоторые плазмиды обладают способностью создавать межклеточные контакты, через которые они и переходят из одной клетки в другую. Образование контактов между донорной и реципиентной клетками обеспечивается конъюгативными свойствами плазмид, а сам перенос ДНК – мобилизационными. Большинство плазмид, которые используются в работах с рекомбинантными ДНК, не обладают конъюгативными функциями и поэтому не могут переходить в реципиентные клетки путем конъюгации. Однако проникновение в клетку некоторых плазмидных векторов все-таки происходит при наличии в этой клетке второй плазмиды, обладающей конъюгативными свойствами. Таким образом, введя в клетку, несущую мобилизуемый плазмидный вектор, плазмиду с конъюгативными функциями, можно трансформировать клетки-реципиенты, с трудом поддающиеся трансформации другими способами.
Опишем в общих чертах используемую для этого стандартную экспериментальную процедуру. Смешивают клетки трех разных штаммов. Когда клетки оказываются в непосредственной близости друг от друга, конъюгативная плазмида, которая в данном случае обладает также мобилизационными свойствами, сама переходит в клетку, содержащую мобилизуемый плазмидный вектор, а затем обеспечивает перенос векторной ДНК в реципиентную клетку. В такой системе реализуются все возможные пути переноса, но подобные штаммы и плазмиды обладают такими генетическими свойствами, чтобы можно было отобрать реципиентную клетку, получившую данный плазмидный вектор. Предположим, что мы имеем три штамма: 1) штамм А, который несет конъюгативную мобилизуемую плазмиду, но не может расти на минимальной питательной среде и чувствителен к антибиотику X; 2) штамм В, который также не может расти на минимальной питательной среде и несет неконъюгативный плазмидный вектор, который имеет ген резистентное™ к антибиотику X; 3) штамм С – рецепиентная клетка-мишень, которая может расти на минимальной среде, не имеет несовместимых плазмид и чувствительна к антибиотику X. После конъюгации клетки непродолжительное время выращивают на полноценной среде без антибиотика X, а затем переносят их на минимальную среду с антибиотиком. В этих условиях могут расти только реципиентные клетки-мишени, которые приобрели плазмидный вектор. Иногда клетка-мишень получает обе плазмиды, однако этот редкий случай можно выявить, если перенести клетки путем перепечатки на минимальную среду и отобрать трансконъюганты, которые способны расти в присуствии антибиотика X, но не могут расти при наличии гена резистентности к другому антибиотику (например, к антибиотику Y), который имеется в конъюгативной плазмиде штамма А. Поскольку для переноса плазмидной ДНК должна произойти-конъюгация между тремя бактериальными штаммами, эта процедура получила название тройного скрещивания.
ЗАКЛЮЧЕНИЕ
Технология рекомбинантных ДНК включает целый набор экспериментальных процедур, благодаря которым удается выделить (клонировать) фрагменты ДНК, содержащие специфические гены. Успех клонирования зависит от возможности воспроизводимо разрезать молекулу ДНК на фрагменты определенного размера. Для точного расщепления ДНК используют рестрицирующие эндонуклеазы типа II. Эти ферменты узнают специфические нуклеотидные последовательности и симметрично разрезают фосфодиэфирные связи в каждой цепи.
Типичный эксперимент по клонированию генов включает следующие этапы. 1. Рестриктазное расщепление ДНК, выделенной из организма, который содержит искомый ген. 2. Обработка вектора для клонирования (обычно плазмидного), который может реплицироваться в клетке-хозяине, теми же рестриктазами, которые использовались для расщепления донорной ДНК. 3. Смешивание этих двух образцов ДНК и сшивание фрагментов ДНК-лигазой фага Т4. 4. Трансформация сшитыми молекулами клеток-хозяев. Амплификация рекомбинантной ДНК в трансформированных клетках.
Для отбора клеток, содержащих рекомбинантную ДНК, используют специальные приемы. Чтобы уменьшить количество кольцевых плазмидных молекул, образующихся при сшивании фрагментов ДНК-лигазой Т4, рестрицированную плазмидную ДНК обрабатывают щелочной фосфатазой, удаляющей 5'-концевые фосфатные группы. Для отбора трансформированных клеток, содержащих гибридные плазмиды, проводят: 1) тестирование на резистентность к определенным антибиотикам или колориметрическую реакцию; 2) иммунологические тесты или выявление специфического белка – продукта клонированного гена; 3) гибридизацию с зондом, комплементарным какому-либо участку искомого гена.
Чтобы иметь возможность клонировать целый ген, донорную ДНК расщепляют лишь частично. При этом получаются фрагменты разной длины, из которых затем создают геномную библиотеку. Для клонирования крупных фрагментов ДНК были сконструированы векторы на основе бактериофагов λ, и Р1, а также плазмиды F.
Для получения фрагментов ДНК, кодирующих эукариотические белки, на очищенной мРНК как на матрице синтезируют комплементарную цепь
ДНК с помощью обратной транскриптазы; эта цепь в свою очередь используется в качестве матрицы для синтеза второй цепи. После ферментативной обработки эту двухцепочечную комплементарную ДНК встраивают в вектор.
Независимо от того, какая именно стратегия клонирования используется, после идентификации клонированной последовательности необходимо показать, что она представляет собой нативный структурный ген.
