Технологический прогресс в любой области науки всегда стимулирует ее дальнейшее развитие. С появлением новых технологий появляется возможность ставить новые эксперименты и облегчается проведение старых. Становление молекулярной биотехнологии как науки обязано целому ряду технологических разработок; многие из них ныне широко применяются как в крупных исследовательских центрах, так и небольших научных коллективах. Теперь не составляет особого труда химически синтезировать одну молекулу ДНК, определить нуклеотидную последовательность другой и амплифицировать с помощью полимеразной цепной реакции третью. Все это стало возможным благодаря той информации, которая была получена в ходе основополагающих исследований как самой ДНК, так и механизма ее репликации. Эти экспериментальные подходы стали неотъемлемой частью молекулярного клонирования – процедуры, позволяющей выделять из ДНК нужные фрагменты, охарактеризовывать их и производить с ними разнообразные манипуляции.
ХИМИЧЕСКИЙ СИНТЕЗ ДНК
С разработкой быстрых и недорогих методов химического синтеза однонепочечных ДНК-фрагментов с заданной нуклеотидной последовательностью методология молекулярного клонирования и характеризации ДНК существенно изменилась. Химически синтезированные олигонуклеотиды можно использовать для конструирования целых генов или их фрагментов, для амплификации специфических фрагментов
ДНК, для направленных мутаций изолированных ДНК, а также в качестве зондов при гибридизации и в качестве линкеров, облегчающих клонирование.
С появлением приборов для автоматического химического синтеза ДНК (ДНК-синтезаторов) получение одноцепочечных олигонуклеотидов длиной ≤50 звеньев стало более или менее рутинной процедурой. Основным компонентом любого ДНК-синтезатора является система клапанов и насосов, с помощью которых в реакционную смесь по строго заданной программе вводятся нуклеотиды и реагенты, обеспечивающие присоединение нужных мономерных единиц к растущей цепи. В отличие от биологического, в ходе химического синтеза ДНК каждый новый нуклеотид можно присоединять к 5'-гидроксильному концу цепи. Все реакции осуществляются последовательно в одной реакционной колонке, а продолжительность каждой из них и время отмывания контролируются с помощью компьютера.
Фосфорамидитный метод
В настоящее время это наиболее распространенный метод химического синтеза ДНК. Исходными строительными блоками в нем являются модифицированные дезоксирибонуклеозиды. Модификация состоит в присоединении к аминогруппам дезоксиаденозина и дезоксицитидина бензольной группы, а к аминогруппе дезоксигуанозина – изобутирильной. Тимидин, у которого отсутствует аминогруппа, не модифицируют. Такая модификация необходима для защиты нуклеозидов от нежелательных побочных реакций при росте цепи. Синтез осуществляют в твердой фазе (растущая цепь ДНК фиксируется на твердом носителе), что позволяет проводить все реакции в одной емкости, легко отмывать после каждого этапа ненужные реагенты и добавлять новые в количестве, обеспечивающем возможно полное протекание реакции.
Этапы многоступенчатого синтеза представлены на рис. 5.1. Первый нуклеозид (азотистое основание + сахар) фиксируют на инертном твердом носителе, обычно это пористые стеклянные шарики с порами одинакового размера. З'-гидроксильная группа первого нуклеозида, который будет 3'-концевым нуклеотидом синтезируемой цепи, прикрепляется к спейсерной молекуле, ковалентно связанной с носителем. Чтобы предотвратить неспецифическое взаимодействие 5'-гидроксильной группы первого нуклеотида до добавления в реакционную смесь второго нуклеотида, ее защищают с помощью диметокситритильной (ДМТ) группы (рис. 5.2). Такую группу содержит каждый присоединяемый к растущей цепи нуклеотид, а кроме того, он несет диизопропиламинную группу, присоединенную к З'-фосфитной группе, которая в свою очередь защищена метальным остатком (рис. 5.3). Такая молекулярная конфигурация и называется фосфорамидитом.
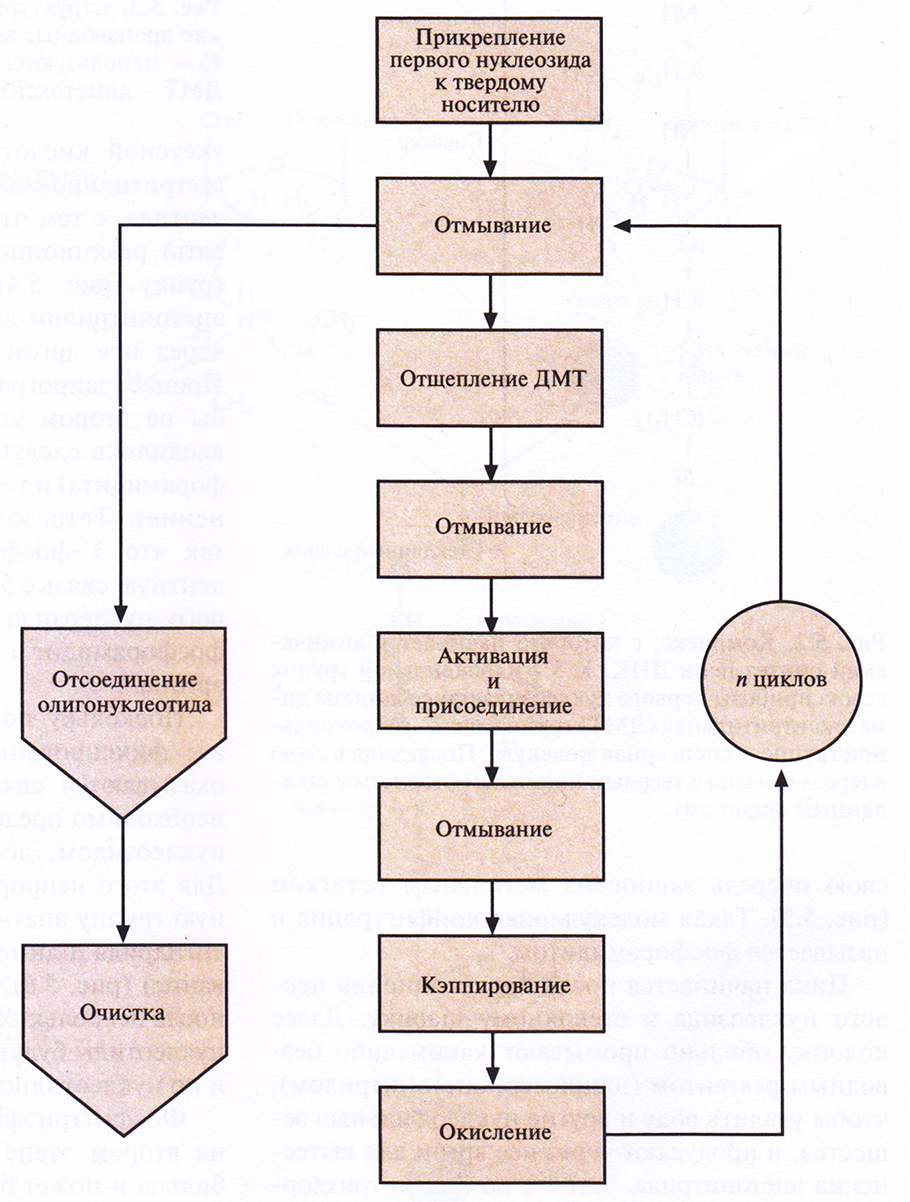
Рис. 5.1. Химический синтез олигонуклеотида. После n циклов образуется одноцепочечный фрагмент ДНК из n + 1 нуклеотида
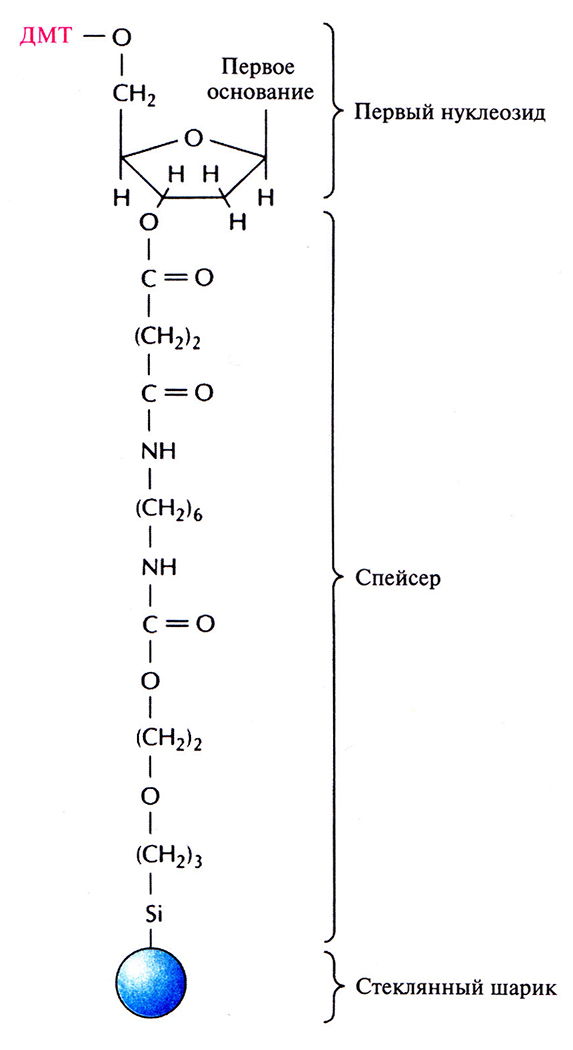
Рис. 5.2. Комплекс, с которого начинается химический синтез цепи ДНК. К 5'-гидроксильной группе дезоксирибозы первого нуклеозида присоединена диметокситритильная (ДМТ) группа, а к З'-гидроксильной группе – спейсерная молекула. Последняя в свою очередь связана с твердым носителем (пористым стеклянным шариком).
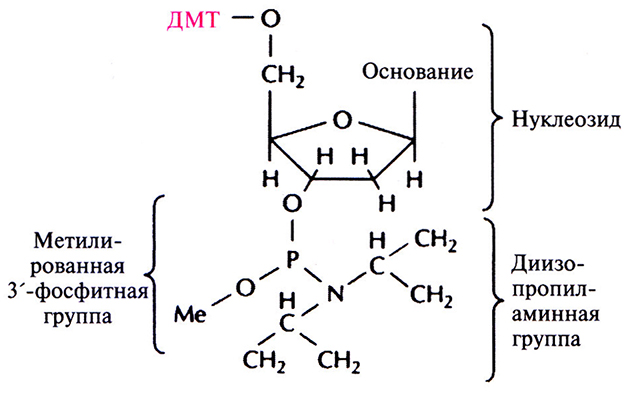
Рис. 5.3. Структурная формула фосфорамидита. Такие производные всех четырех оснований – А, Т, G и С – используются для химического синтеза ДНК. ДМТ – диметокситритил, Me – метальная группа.
Цикл начинается после присоединения первого нуклеозида к стекляному шарику. Далее колонку обильно промывают каким-либо безводным реагентом (например, ацетонитрилом), чтобы удалить воду и другие нуклеофильные вещества, и продувают через нее аргон для вытеснения ацетонитрила. Затем с помощью трихлор-уксусной кислоты (ТХУ) отщепляют 5'-ДМТ (детритилирование) от присоединенного нуклеотида, с тем чтобы высвободить (экспонировать) реакционноспособную 5'-гидроксильную группу (рис. 5.4). Колонку вновь промывают ацетонитрилом для удаления ТХУ и продувают через нее аргон для удаления ацетонитрила. Процесс запрограммирован таким образом, чтобы на втором этапе в колонку одновременно вводились следующий нуклеозид (в виде фосфорамидита) и тетразол (активация и присоединение). Тетразол активирует фосфорамидит, так что З'-фосфитная группа образует ковалентную связь с 5'-гидроксильной группой первого нуклеозида (рис. 5.5). Невключившийся фосфорамидит и тетразол удаляют продуванием аргона.
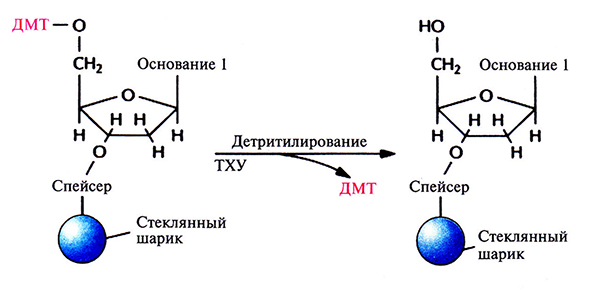
Рис. 5.4. Детритилирование – отщепление 5'-диметокситритильной (ДМТ) группы с помощью трихлоруксусной кислоты (ТХУ).
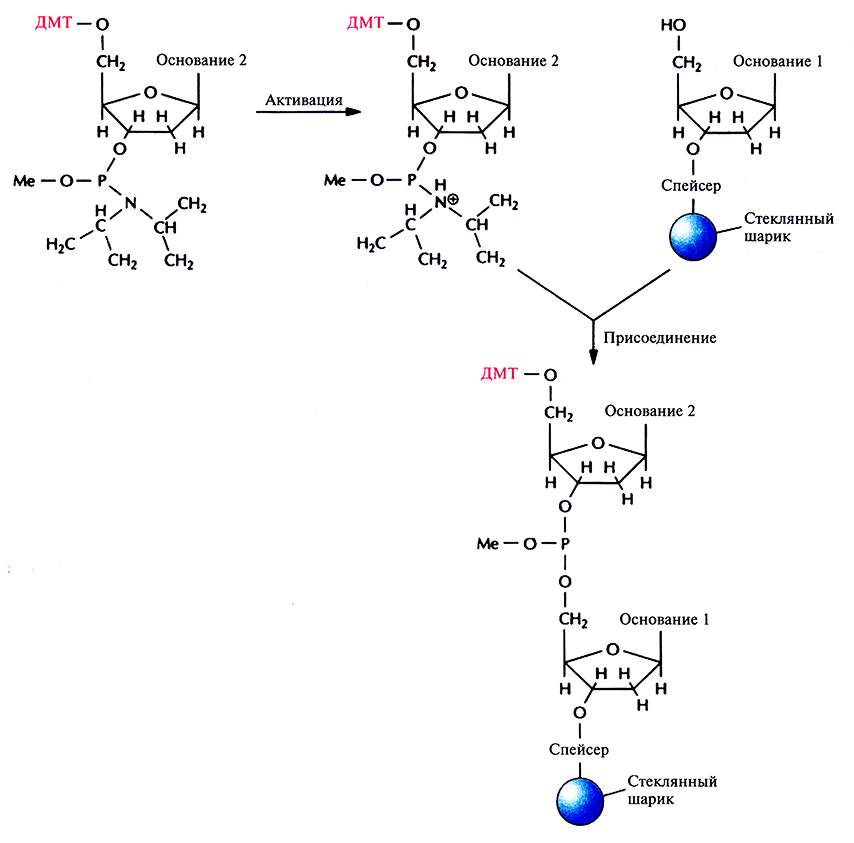
Рис. 5.5. Активация и присоединение. З'-фосфитная группа активированного фосфорамидита образует ковалентную связь с 5'-гидроксильной группой фиксированного на стеклянном шарике детритилированного нуклеозида. ДМТ – диметокситритильная группа, Me – метильная группа.
Поскольку по окончании первого этапа не все фиксированные на носителе нуклеозиды оказываются связанными с фосфорамидитом, необходимо предотвратить их взаимодействие с нуклеозидом, добавленным на втором этапе. Для этого непрореагировавшую 5'-гидроксильную группу ацетилируют с помощью уксусного ангидрида и диметиламинопиридина (кэппирование) (рис. 5.6). Если этого не сделать, то уже после нескольких циклов синтезируемые олигонуклеотиды будут различаться как по длине, так и по нуклеотидной последовательности.
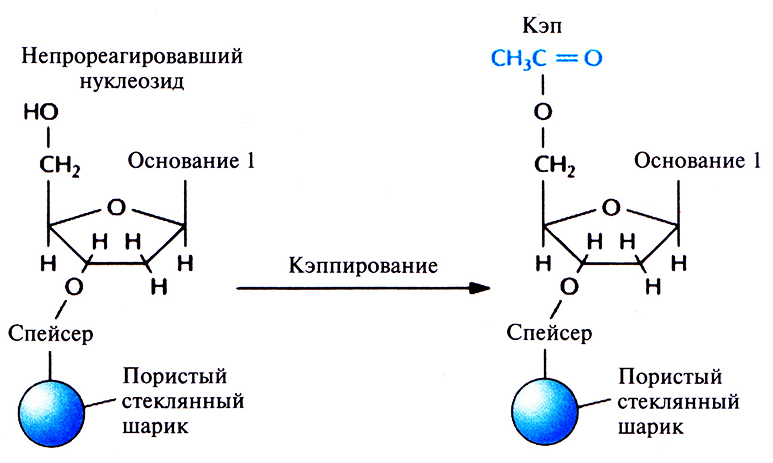
Рис. 5.6. Кэппирование. Свободные 5'-гидроксильные группы непрореагировавших в первом цикле детритилированных нуклеозидов ацетилируют, чтобы предотвратить их участие в следующем цикле.
Фосфиттриэфирная связь, образовавшаяся на втором этапе между нуклеотидами, нестабильна и может разорваться в присутствии кислоты или щелочи. Поэтому фосфиттриэфир окисляют с помощью йодной смеси до более стабильного пятивалентного фосфаттриэфира (рис. 5.7). Затем промывают колонку и повторяют весь цикл (детритилирование, активация и присоединение, кэппирование, окисление; рис. 5.1). Все описанные операции проводят до тех пор, пока к растущей цепи в соответствии с программой не присоединится последний нуклеозид. Синтезированные олигонуклеотиды связаны со стеклянными шариками; каждый фосфаттриэфир несет метальную группу; каждый гуанин, цитозин и аденин содержит защищенную аминогруппу, а на 5'-конце последнего нуклеотида находится ДМТ-группа.
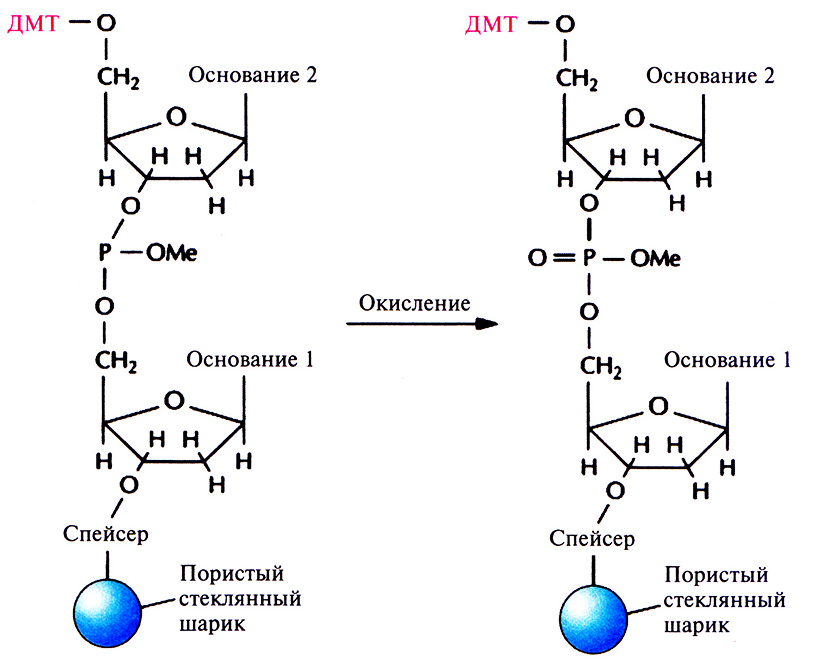
Рис. 5.7. Окисление. Фосфиттриэфир окисляется до пятивалентного фосфаттриэфира, что приводит к стабилизации фосфодиэфирной связи и делает ее более устойчивой к действию кислот и шелочей. ДМТ – диметокситритильная группа, Me – метильная группа.ским методом. Эту реакцию можно проводить и тогда, когда олигонуклеотид еще связан с носителем, но после детритилирования.
Метильные группы удаляют с помощью химической обработки непосредственно в реакционной колонке. Затем отсоединяют олигонуклеотиды от спейсерной молекулы вместе с З'-гидроксильным концом и элюируют их из колонки; далее последовательно удаляют бензоильные, изобутирильные и ДМТ-группы. 5'-конец цепи фосфорилируют ферментативным (полинуклеотидкиназа Т4+АТР) или химическим методом. Эту реакцию можно проводить и тогда, когда олигонуклеотид еще связан с носителем, но после детритилирования.
Чтобы выход продукта был достаточно высок, эффективность присоединения нуклеотидов на каждом этапе должна быть не ниже 98%. Эффективность контролируют спектрофотометрическими методами, определяя количество удаляемых тритильных групп. Если, например, при синтезе 20-членного олигонуклеотида эффективность каждого цикла равна 99%, то 82% (т. е. 0,9920 • 100) олигонуклеотидов будут иметь именно такую длину. Если же синтезируется 60-членный олигонуклеотид, то при той же эффективности только 55% олигонуклеотидов будут содержать по 60 нуклеотидов. А если средняя эффективность цикла не превышает 98%, то доля олигонуклеотидов заданной длины будет гораздо ниже (табл. 5.1). Фирмы – изготовители коммерческих ДНК-синтезаторов обычно гарантируют среднее значение эффективности присоединения 98%. Но для этого необходимо использовать реагенты и химикаты очень высокой степени чистоты, что не всегда удается выполнить. Как правило, реальная эффективность присоединения составляет 95%, хотя иногда удается достичь и 99%-ной эффективности Чтобы получить олигонуклеотиды заданной длины, первичные продукты большинства химических синтезов необходимо очистить с помощью либо высокоэффетивной жидкостной хроматографии под высоким давлением с обращенной фазой, либо электрофореза в полиакриламидном геле. Поскольку все «неудачные» последовательности короче, чем тот олигонуклеотид, который хотят получить, сделать это нетрудно.
Таблица 5.1. Средний выход олигонуклеотидов заданной длины
(и) при разных значениях средней эффективности цикла

Применение синтезированных олигонуклеотидов
Олигонуклеотиды, синтезированные химическими методами, находят широкое применение в молекулярной биотехнологии. Их используют в качестве зондов при ДНК-гибридизации, линкеров, соединяющих разные молекулы ДНК в экспериментах по клонированию, праймеров при секвенировании ДНК или осуществлении сайт-специфического мутагенеза клонированных генов-мишеней.
1. Нуклеотидную последовательность специфических олигонуклеотидных зондов (длиной 20–40 звеньев) находят из данных об аминокислотной последовательности соответствующих белков.
2. Для получения линкеров синтезируют олигомеры, которые представляют собой палиндромные одноцепочечные нуклеотидные последовательности, спаривающиеся (гибридизующиеся) между собой. Линкеры содержат сайты узнавания для рестрицирующих эндонуклеаз, что позволяет осуществлять с их помощью клонирование фрагментов ДНК (рис. 5.8, А и Б). Короткий дуплекс длиной 6–12 пар нуклеотидов лигируют по тупым концам с ДНК-мишенью (обычно кДНК). Разрезают новую молекулу нужной рестрицирующей эндонуклеазой и получают фрагменты с выступающими одноцепочечными концами (липкими концами), с помощью которых встраивают ДНК-мишень в соответствующий вектор. Прежде чем проводить встраивание, рестрицированную смесь фракционируют для отделения ДНК с липкими концами от лишних линкерных молекул. Вектор тоже обрабатывают рестриктазой, отжигают его с фрагментами ДНК с липкими концами и сшивают с помощью ДНК-лигазы фага Т4. ДНК-мишень не должна содержать сайтов рестрикции, присутствующих в линкерной последовательности, в противном случае она также будет расщепляться ферментом.
3. Один из вариантов линкерных последовательностей, так называемые «адаптеры», часто содержат сайты для двух и более рестрицирующих эндонуклеаз (рис. 5.8, В). С их помощью можно встраивать кДНК в вектор лигированием по тупым концам, а затем вырезать, используя другую рестриктазу. Адаптор, изображенный на рис. 5.8, В, встраивают в BamHll-сайт вектора перед включением ДНК-мишени в Smal-сайт по тупым концам. После клонирования кДНК вырезают из вектора с помощью рестриктазы BamHll. В этом случае вектор не должен содержать Smal-сайтов, и ни вектор, ни кДНК не должны нести BamHll-сайтов.
4. Одноцепочечные олигонуклеотиды из ~17–24 звеньев используют в качестве праймеров при секвенировании ДНК и проведении ПЦР.
5. Одноцепочечные олигонуклеотиды используют в качестве праймеров для сайт-специфического мутагенеза in vitro.
6. Необходимость в химическом синтезе нуклеотидной последовательности, кодирующей какой-то конкретный белок, может возникнуть тогда, когда клонирование соответствующего гена затруднено. При этом нуклеотидную последовательность гена находят из данных об аминокислотной последовательности белка. К химическому синтезу прибегают и тогда, когда кодоны, из которых состоит данный ген, плохо считываются организмом-хозяином, и уровень трансляции оказывается очень низким. В таком случае можно синтезировать ген с таким набором кодонов (оптимизация кодонов), при котором аминокислотная последовательность кодируемого белка остается прежней, а кодоны считываются хозяйским организмом более эффективно.
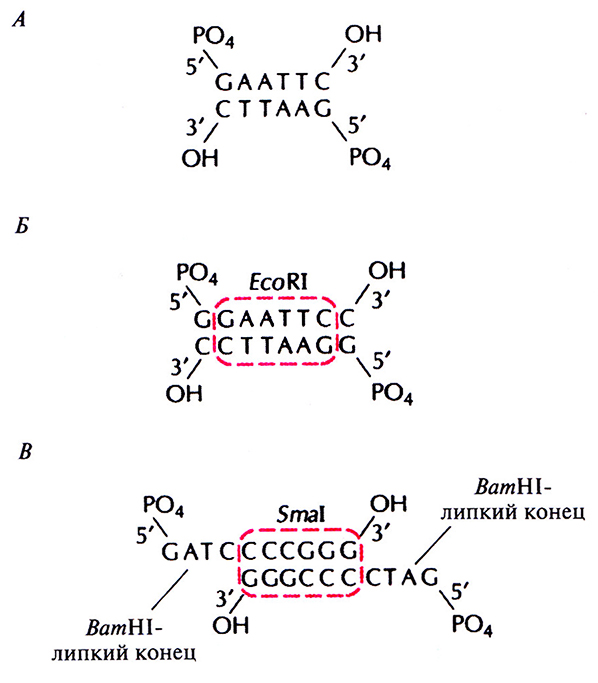
Рис. 5.8. Типичные линкеры и адаптор. А. EcoRI-линкер, состоящий из 6 пар нуклеотидов. Б. EcoRl-линкер из 8 пар нуклеотидов. В. BamHl-Smal-адаптор с BamHl-липкими концами и сайтом узнавания для Smal.
Синтез генов
Если химически синтезированную двухцепочечную ДНК предполагается использовать в качестве гена или его фрагмента, то каждую из ее цепей синтезируют отдельно. Получить короткие гены (60–80 п. н.) технически несложно: для этого синтезируют комплементарные цепи и затем отжигают их. В случае крупных генов (>300 п. н.) приходится применять специальные стратегии, поскольку эффективность каждого цикла химического синтеза никогда не достигает 100%. Например, если ген состоит из 999 пар нуклеотидов, а эффективность каждого цикла равна 99%, то доля полноразмерных одноцепочечных ДНК по окончании процесса составит не более 0,004%. Чтобы решить эту проблему, синтетические (двухцепочечные) гены собирают из модулей – (одноцепочечных) фрагментов длиной от 20 до 100 нуклеотидов.
Один из способов конструирования синтетического гена заключается в получении набора олигонуклеотидов длиной 20–60 нуклеотидов каждый с перекрывающимися концами. Нуклеотидные последовательности цепей задают так, чтобы после отжига концевые сегменты гена имели тупые концы. Каждый внутренний сегмент имеет выступающие 3'- и 5'-концы, комплементарные таковым соседнего сегмента (рис. 5.9). После сборки гена остается сшить одноцепочечные разрывы с помощью ДНК-лигазы Т4. Синтетические гены могут быть сконструированы так, чтобы помимо белок-кодирующих последовательностей они содержали концевые участки, обеспечивающие их встраивание в клонирующий вектор (сайты для рестрицируюших эндонуклеаз), а также, если это необходимо, сигнальные последовательности для правильной инициации и терминации транскрипции и трансляции.
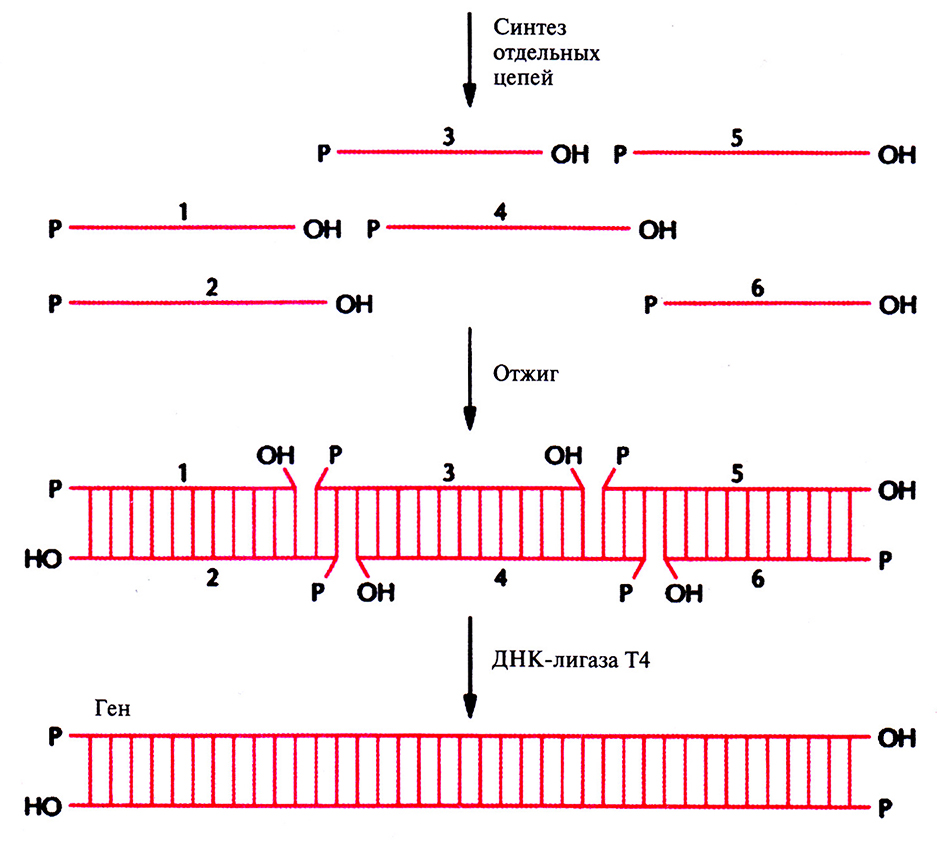
Рис. 5.9. Сборка синтетического гена из коротких олигонуклеотидов. Синтезируют отдельные олигонуклеотиды длиной от 20 до 60 звеньев каждый с такими нуклеотидными последовательностями, чтобы при отжиге из них образовалась двухиепочечная молекула. Оставшиеся одноцепочечные разрывы сшивают с помощью ДНК-лигазы Т4.
Для получения полноразмерных генов другим способом тоже вначале синтезируют специфический набор перекрывающихся олигонуклеотидов длиной от 40 до 100 звеньев. При их отжиге происходит спаривание 6–10 3'- и 5'-концевых взаимно комплементарных нуклеотидов, а между ними остаются большие бреши. Протяженность спаренных участков достаточно велика, чтобы стабилизировать всю структуру. Бреши заполняют ферментативным путем с помощью ДНК-полимеразы I Escherichia coli, использующей З'-гидроксильные группы для инициации репликации и одноцепочечные участки в качестве матрицы. Оставшиеся одноцепочечные разрывы сшивают с помощью ДНК-лигазы Т4 (рис. 5.10).
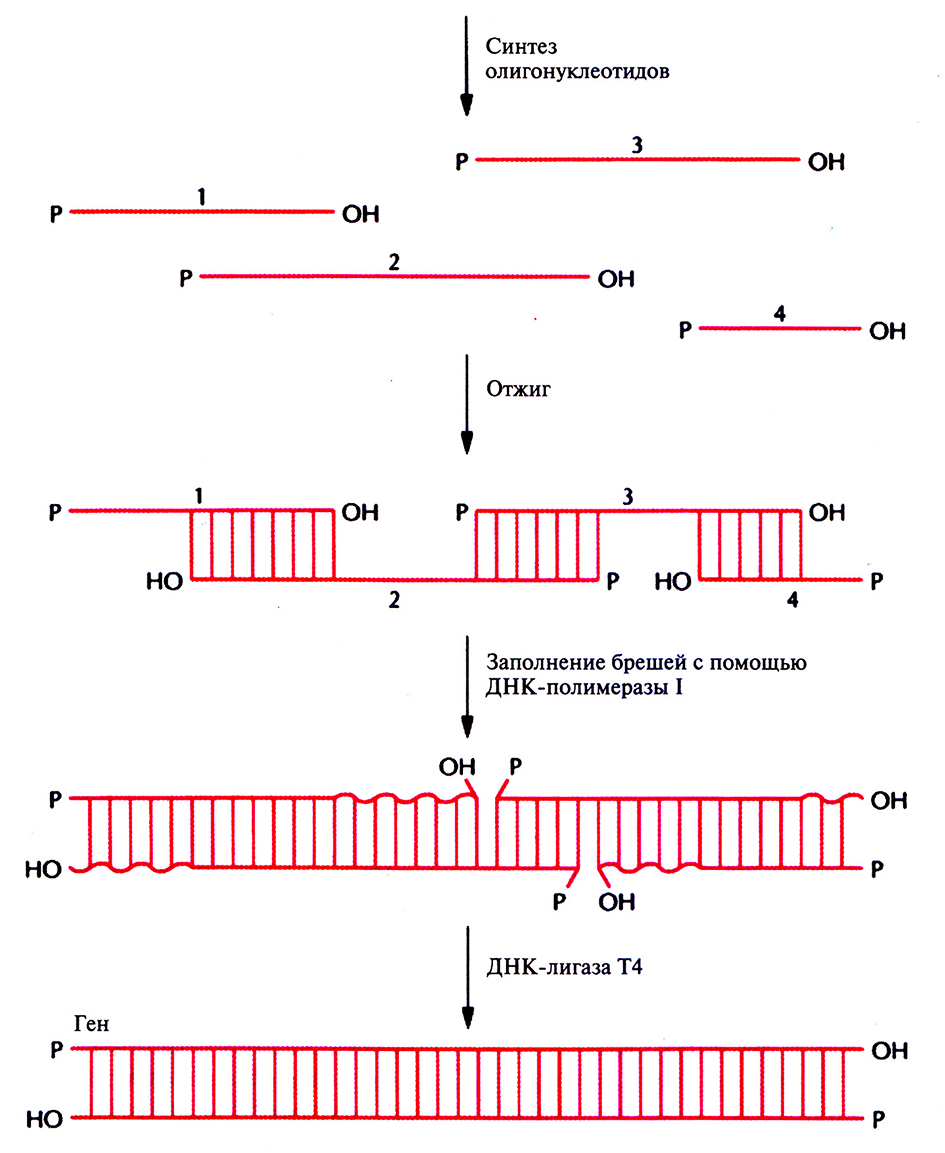
Рис. 5.10. Сборка протяженного гена in vitro с участием ферментов. Вначале химическими методами синтезируют отдельные олигонуклеотиды с такими нуклеотидными последовательностями, чтобы при отжиге между ними образовывались спаренные участки длиной 6–10 пар нуклеотидов. Оставшиеся между ними бреши заполняют с помощью ДНК-полимеразы I Е. coli, а одноцепочечные разрывы сшивают ДНК-лигазой Т4.
Более протяженные гены (>1000 п. н.) обычно собирают из двухцепочечных фрагментов, каждый из которых в свою очередь состоит из 4–6 перекрывающихся олигонуклеотидов (от 20 до 60 п. н. каждый). Если после синтеза и отжига образуется достаточное количество фрагментов, то их просто соединяют друг с другом. В противном случае каждый фрагмент клонируют и амплифицируют. Двухцепочечные фрагменты последовательно соединяют друг с другом до образования полноразмерного гена. Чтобы гарантировать правильность нуклеотидноq последовательности химически синтезированного гена, секвенируют каждый двухцепочечный фрагмент, а затем и весь ген.
ВАЖНАЯ ВЕХА
Секвенирование ДНК с помощью терминирующих дидезоксинуклеотидов
F. Sanger, S. Nicklen, A. R. Coulson Proc. Natl. Acad. Sci. USA 74: 5463-5467, 1977
Специфическая ферментативная амплификация ДНК in vitro: полимеразная цепная реакция
К. В. Mullis, F. A. Faloona, S. J. Scharf, R. К. Saiki, G. Т. Horn, H. A. Erich Cold Spring Harbor Symp. Quant. Biol. 51: 263-273, 1986
Создание новых методов – это необходимая предпосылка развития любой отрасли науки. Они позволяют получать недоступную прежде информацию, что в свою очередь приводит к более глубокому пониманию сути наблюдаемых явлений и стимулирует дальнейшие исследования, порождающие новые открытия. Что касается молекулярной биотехнологии, то ее основой стали такие мощные методы, как секвенирование ДНК и ПЦР.
Определение нуклеотидной последовательности ДНК методом ферментативного копирования с остановкой удлинения цепи, осуществляемого ДНК-полимеразой, – быстрый, относительно простой, недорогой и надежный метод. Помимо того, что нуклеотидная последовательность фрагмента ДНК является его исчерпывающей характеристикой на молекулярном уровне, она позволяет также идентифицировать его кодирующую область, подобрать потенциальные праймеры для ПЦР, выявить мутационные изменения в гене. До появления в 1977 г. дидезокси метода Сангера для Секвенирования ДНК использовали метод сайт-специфического химического расщепления цепи (А. М. Maxam, W. Gilbert, Proc. Natl. Acad. Sci. USA 74: 560-564, 1977). А еще раньше секвенирование нуклеиновых кислот сводилось к определению нуклеотидных последовательностей РНК. Для этого нужный фрагмент ДНК сначала транскрибировали в РНК с помощью РНК-полимеразы, а затем определяли нуклеотидную последовательность последней. Процедура была весьма сложной и длительной и состояла в следующем: радиоактивно меченную РНК обрабатывали различными рибонуклеазами, затем осуществляли хроматографическое разделение образовавшихся продуктов, повторно обрабатывали их ферментами, проводили щелочной гидролиз продуктов второго расщепления, осуществляли хроматографическое разделение продуктов гидролиза, определяли очередность олигонуклеотидов, основываясь на перекрывании их концевых участков, и воссоздавали исходную молекулу. С появлением дидезокси-метода эта процедура практически перестала использоваться. Теперь секвенируют не саму РНК, а ДНК, синтезированную на РНК как на матрице с помощью обратной транскриптазы, и применяют не метод Максама и Гилберта, а метод Сангера, появившийся после того, как была создана система клонирования на основе фага М13. Возможность прямого секвенирования ДНК произвела настоящую революцию в исследовании молекулярных основ различных болезней человека и в разработке методов их диагностики и лечения.
Огромное влияние на многие области исследований, в том числе и на молекулярную биотехнологию, оказала разработка метода ПЦР (Kary Mullis; U.S. patent 4,683,202). С возможностью получения больших количеств ДНК амплификацией сегментов клонированной или геномной ДНК была решена проблема клонирования ДНК-копий редких молекул мРНК, скрининга геномных библиотек, выявления генных мутаций, физического картирования хромосом и т. д. Первым практическим применением ПЦР было создание тест-системы для диагностики серповидноклеточной анемии (Saiki et al., Science 230: 1350-1354, 1985). ПЦР – это уникальная методика, равных которой нет среди других хорошо известных методов. Начиная с 1986 г. с ее помощью выполнено более 10 000 исследований, и несмотря на их огромное разнообразие, перспективы использования ПЦР представляются еще более впечатляющими.
МЕТОДЫ СЕКВЕНИРОВАНИЯ ДНК
Исчерпывающую информацию о молекуле ДНК можно получить, только определив ее нуклеотидную последовательность. Так, секвенировав ген, часто удается установить его функцию, сравнив его нуклеотидную последовательность с таковыми для генов, функция которых уже известна. Без данных о нуклеотидной последовательности невозможно проводить исследования по молекулярному клонированию. Секвенирование того или иного фрагмента ДНК можно провести либо химическим методом, разработанным А. Максамом и В. Гилбертом, либо ферментативным, предложенным Ф. Сангером, но в настоящее время наиболее широко используется так называемый дидезоксинуклеотидный метод.
Дидезоксинуклеотидный метод секвенирзвания ДНК
Дидезоксинуклеотид – это полученный искусственным путем нуклеотид, лишенный 2'- и 3'-гидроксильных групп при углеродных атомах сахарного кольна (рис. 5.11, А). У дезоксинуклеотида, входящего в норме в состав ДНК, отсутствует только 2'-гидроксильная группа (рис. 5.11, Б). Удлинение цепи во время репликации ДНК происходит в результате присоединения очередного нуклеозидтрифосфата к З'-гидроксильной группе последнего нуклеотида растущей цепи (рис. 5.12). И если таким очередным присоединяемым звеном является дидезоксинуклеотид, то синтез ДНК останавливается, поскольку следующий нуклеотид не может образовать фосфодиэфирную связь (рис. 5.13). Остановка синтеза ДНК – это ключевой этап дидезоксиметода, но чтобы осуществить секвенирование в полном объеме, необходимо выполнить целый ряд условий.
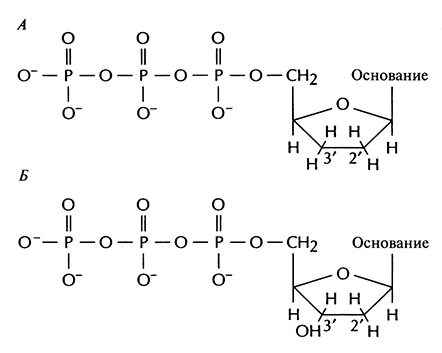
Рис. 5.11. А. Дидезоксинуклеотид (отсутствуют 2'- и З'-гидроксильные группы в кольце). Б. Дезоксинуклеотид (отсутствует только 2 -гидроксильная группа).
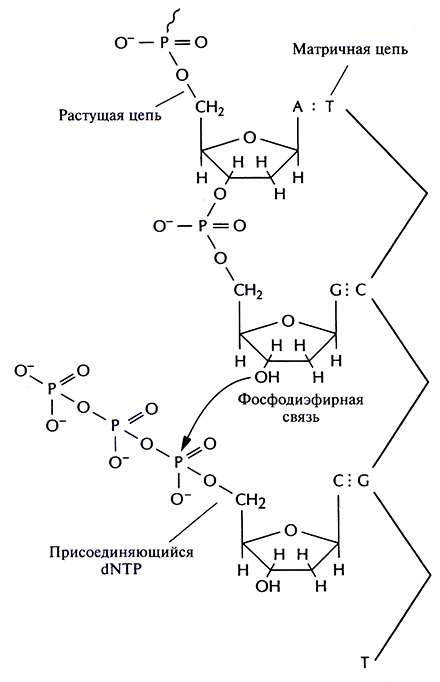
Рис. 5.12. Синтез ДНК в обычных условиях. Очередной дезоксирибонуклеотид (дезоксирибонуклеозидтрифосфат; dNTP) спаривается с комплементарным нуклеотидом матричной цепи. Между З'-гидроксильной группой последнего нуклеотида в растущей цепи и а-фосфатной группой присоединяемого нуклеотида образуется фосфодиэфирная связь.
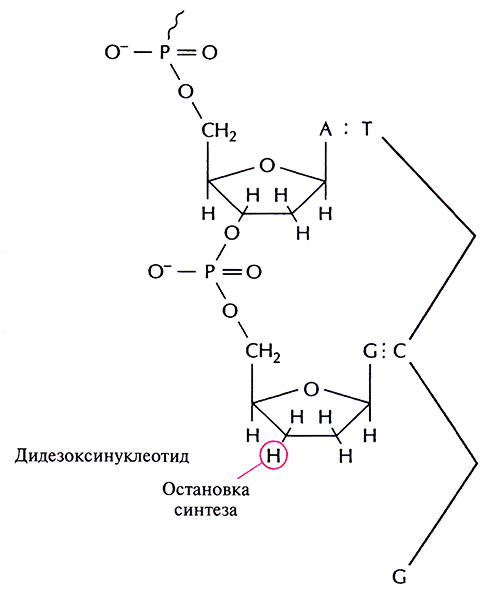
Рис. 5.13. Остановка синтеза ДНК после присоединения дидезоксинуклеотида к концу растущей цепи. Фосфодиэфирная связь между концевым дидезоксинуклеотидом и следующим нуклеотидом не может образоваться из-за отсутствия З'-гидроксильной группы.
Первый шаг стандартной процедуры дидезоксисеквенирования состоит в гибридизации синтетического олигонуклеотида длиной 17–20 звеньев со специфическим участком одной из цепей клонирующего вектора, соседствующим со вставкой. Этот олигонуклеотид является праймером, поставляющим З'-гидроксильную группу для инициации синтеза. Раствор с праймером распределяют по четырем пробиркам, в каждой из которых находятся четыре дезоксинуклеотида, dATP, dCTP, dGTP и dTTP (один из них – изотопно меченный), и один из четырех дидезоксинуклеотидов (ddATP, ddCTP, ddGTP или ddTTP). Концентрацию каждого дидезоксинуклеотида подбирают таким образом, чтобы он оказался включенным по всем позициям в смеси растущих цепей, а не только в первой встретившейся ему позиции. (Напомним, что после присоединения дидезоксинуклеотида рост цепи сразу останавливается, поэтому каждая цепь оканчивается 3'-дидезоксинуклеотид ом.) По окончании ферментативного синтеза при участии ДНК-полимеразы в каждой пробирке оказывается уникальный набор олигонуклеотидов, каждый из которых содержит праймерную последовательность (рис. 5.14).
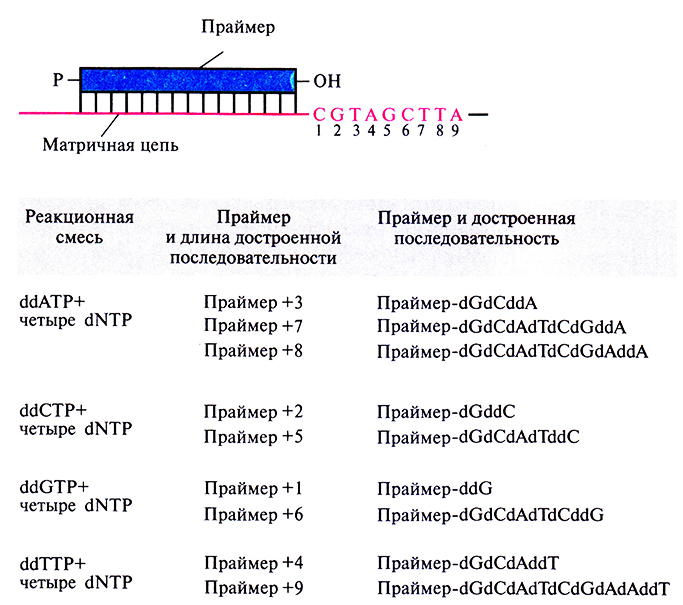
Рис. 5.14. Удлинение праймера при синтезе цепи в присутствии дидезоксинуклеотидов. В каждой из четырех пробирок образуется уникальный набор олигонуклеотидов разной длины, включающих праймерную последовательность. При этом образуется и какое-то число полноразмерных молекул ДНК. dNTP – дезоксинуклеозидтрифосфат.
Далее в пробирки добавляют формамид, чтобы обеспечить расхождение цепей, и проводят электрофорез в полиакриламидном геле на четырех дорожках (по числу пробирок). Это позволяет разделить одноцепочечные фрагменты ДНК, даже если они различаются по длине всего на один нуклеотид. На радиоавтографе обнаруживается набор полос, отвечающих меченым фрагментам ДНК, сопоставление которых позволяет прямо «прочитать» нуклеотидную последовательность секвенируемого сегмента ДНК. В примере, приведенном на рис. 5.15, первые шесть нуклеотидов этого сегмента, начиная с 5'-конца, – это AGCTGC. Самая «быстрая» полоса (радиоактивно меченный фрагмент в самом низу геля) соответствует самому короткому фрагменту и находится в дорожке ddATP; следующие полосы располагаются соответственно в дорожках ddGTP, ddCTP, ddTTP и т. д. На большинстве радиоавтографов четко различаются от 250 до 350 полос. Праймерная последовательность находится на фиксированном расстоянии (10–20 нуклеотидов) от того сайта, по которому встроена клонированная ДНК, что позволяет легко распознать начало клонированного фрагмента.
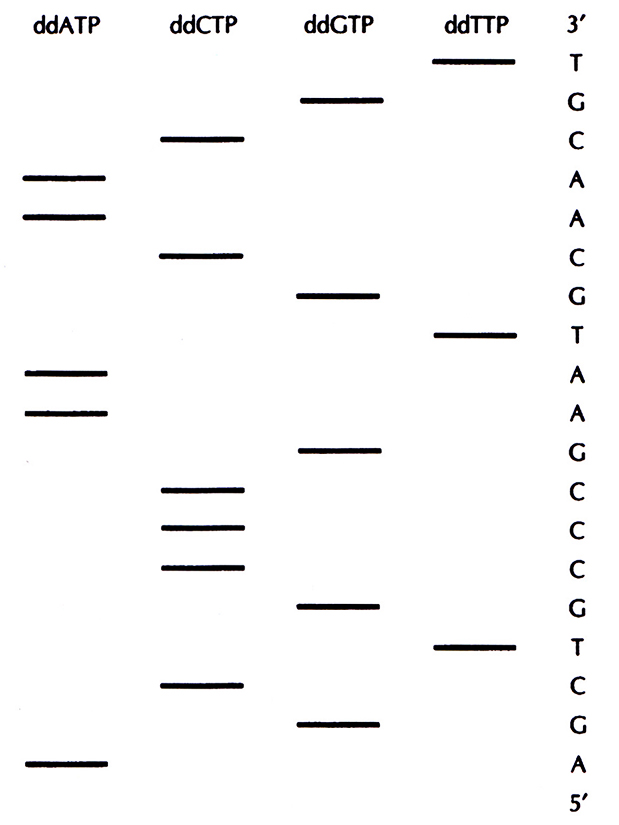
Рис. 5.15. Схематическое изображение радиоавтографа, получающегося при секвенировании ДНК с помощью дидезокси-метода. в каждую из лунок вносили содержимое одной из четырех пробирок, в которых находился один из дидезоксинуклеотидов: ddATP, ddCTP, ddGTP или ddTTP. Нуклеотидная последовательность считывается с радиоавтографа снизу вверх. На рисунке она приведена справа.
Секвенирование ДНК с помощью вектора на основе фага М13
Для определения нуклеотидной последовательности клонированных ДНК используются разные подходы. Один из первых основывался на применении фага Ml3 E. coli в качестве вектора. ДНК этого фага представляет собой одноцепочечную кольцевую молекулу. Когда им инфицируют Е. coli, сначала образуется двухцепочечная репликативная форма фаговой ДНК, а одноцепочечные кольцевые молекулы, которые затем упаковываются в вирионы, синтезируются на этой двухцепочечной молекуле как на матрице. Клетки, инфицированные М13, не подвергаются лизису; в них непрерывно образуются новые одноцепочечные молекулы ДНК М13, которые, проходя через клеточную мембрану, одеваются белковой оболочкой и выходят в окружающую среду. ДНК М13 содержит несущественную часть, которую можно заменить нужным фрагментом ДНК; при этом инфекционность рекомбинантных вирусных частиц сохранится. МП-система имеет следующие преимущества: выделенная двухцепочечная репликативная форма может функционировать как плазмида, а одно-цепочечная фаговая ДНК – использоваться в качестве матрицы для секвенирования ДНК.
Все это позволяет использовать фаг М13 как комбинированную систему для клонирования и секвенирования ДНК. Обычно нужный фрагмент ДНК длиной примерно 500 п. н. встраивают в полилинкер, который является частью клонированного в РФ-ДНК фага М13 модифицированного гена lacZ'. Рекомбинантной вирусной ДНК трансформируют компетентные клетки Е. colin высевают их на чашки со средой, содержащей субстрат X-Gal. При его гидролизе β-галактозидазой образуется продукт, имеющий синюю окраску. На чашках появляются белые (бесцветные) и синие колонии. Первые отвечают клеткам, инфицированным фагом М13 со вставкой, нарушившей рамку считывания гена lacZ', вторые – клеткам, инфицированным фагом М13 с функциональным геном lacZ', не несущим вставки. Из белых колоний выделяют фаговые частицы, а из них – одноцепочечную ДНК со вставкой (рис. 5.16). Для секвенирования последней отжигают выделенную ДНК с праймером, который гибридизуется с последовательностью вблизи вставки, затем проводят дидезокси-секвенирование, электрофорез и радиоавтографию и «прочитывают» нуклеотидную последовательность вставки.
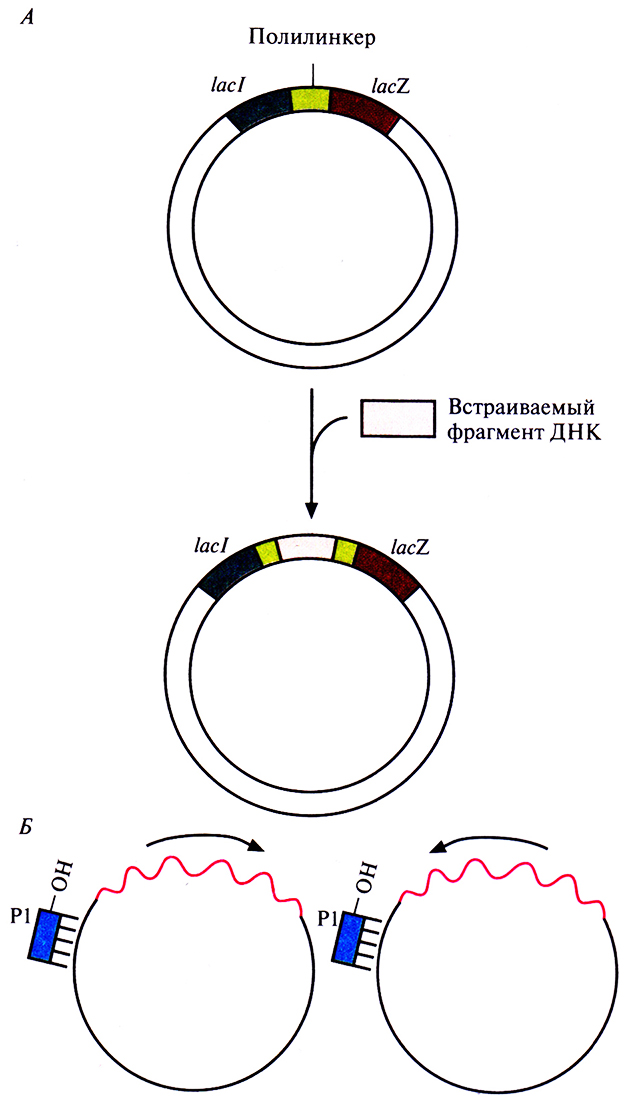
Рис. 5.16. Использование бактериофага М13 для клонирования и секвенирования. А. Встраивание фрагмента ДНК в двухцепочечную репликативную форму ДНК М13. Б. Секвенирование комплементарных цепей клонированного фрагмента ДНК с помощью одного и того же праймера (Р1). Стрелками показана ориентация вставки в векторе.
Для секвенирования крупных фрагментов ДНК (примерно 2000 п. н.) используют другие стратегии. Одна из них состоит в следующем: встраивают этот фрагмент в соответствующий плазмидный вектор и строят его подробную рестрикционную карту. Идентифицируют перекрывающиеся фрагменты вставки длиной от 100 до 500 п. н., субклонируют каждый из них в ДНК М13, секвенируют и воссоздают нуклеотидную последовательность всего исходного фрагмента. Чтобы быть уверенным в правильности полученного результата и идентификации какого-либо нуклеотида, необходимо секвенировать обе цепи по нескольку раз. Секвенирование обеих цепей облегчается тем, что каждый из субклонированных фрагментов исходной ДНК может быть встроен в ДНК М13 в противоположных ориентациях. В результате в одном случае праймер будет иницировать синтез первой цепи, а в другом – второй.
Праймер-опосредованная прогулка
(«Блуждающая затравка»)
Для секвенирования очень длинных фрагментов ДНК (>5000 п. н.) описанный выше подход уже не может быть использован, поскольку число М13-векторов, содержащих перекрывающиеся субклонированные последовательности, значительно увеличивается. Чтобы решить эту задачу, были разработаны методы секвенирования двухцепочечных плазмидных ДНК, не требующие субклонирования. Плазмидную ДНК, содержащую нужную вставку, выделяют и отжигают с синтетическим олигонуклеотидным праймером, который гибридизуется с последовательностью в одной из цепей векторной ДНК, находящейся вблизи вставки. Затем осуществляют дидезокси-секвенирование, позволяющее идентифицировать первые 250–350 нуклеотидов вставки. Исходя из этих данных синтезируют второй олигонуклеотидный праймер, комплементарный сегменту вставки, отстоящему примерно на 300 нуклеотидов от места связывания первого праймера, и секвенируют следующие 250–350 нуклеотидов. Аналогичным образом синтезируют третий праймер и определяют нуклеотидную последовательность следующих 250–350 нуклеотидов (рис. 5.17). Эту процедуру, называемую праймер-опосредованной прогулкой, продолжают до тех пор, пока не секвенируют весь фрагмент. Аналогичным образом секвенируют вторую цепь, начиная с праймера, который гибридизуется с этой цепью вблизи вставки.
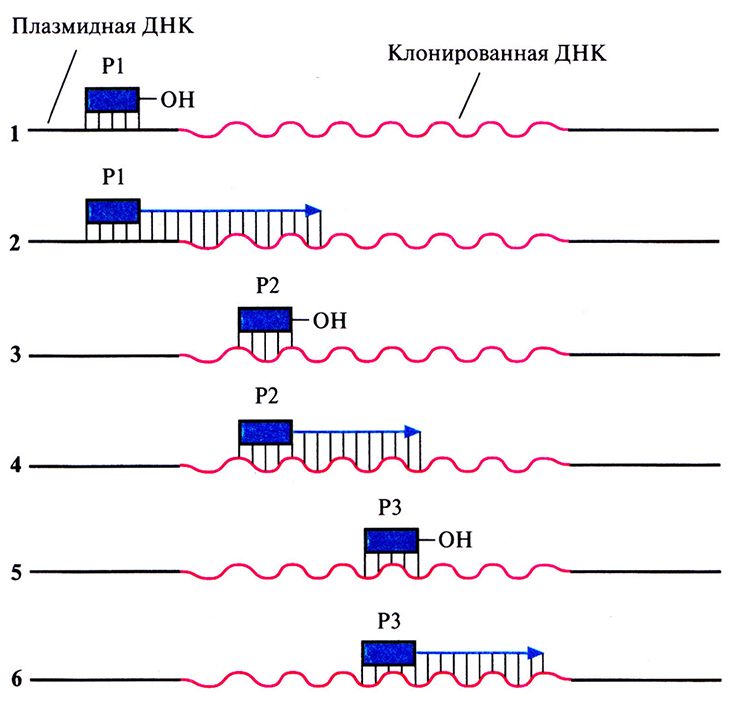
Рис. 5.17. Секвенирование ДНК методом праймер-опосредованной прогулки.
1. Инициация синтеза цепи ДНК с помощью праймера (Р1), комплементарного участку плазмиды, находящемуся вблизи вставки.
2. Секвенирование фрагмента клонированной ДНК длиной 250–350 нуклеотидов.
3. Подбор второго праймера, комплементарного концевому участку уже секвенированной последовательности длиной примерно 20 нуклеотидов.
4. Секвенирование следующего сегмента клонированной ДНК с помощью второго праймера (Р2).
5. Подбор третьего праймера, комплементарного концевому участку этого сегмента размером 20 нуклеотидов.
6. Третий праймер (РЗ) используется для секвенирования следующего сегмента клонированной ДНК. Эту процедуру продолжают до тех пор, пока не будет секвенирована вся вставка.
К сожалению, в результате ошибочного спаривания праймера заданной длины с более чем одним участком внутри вставки могут быть получены неоднозначные результаты. Чтобы избежать этого, используют праймеры длиной не менее 24 нуклеотидов и стараются строго соблюдать условия отжига. Именно таким образом были секвенированы фрагменты ДНК, клонированные в бактериофаге λ (~20 т. п. н.) или в космидном векторе (~40 т. п. н.).
Некоторые этапы этого процесса недавно были автоматизированы. Это позволило проводить рутинное секвенирование фрагментов длиной несколько десятков тысяч пар нуклеотидов. Во многих случаях праймеры, добавляемые в реакционную смесь в разных пробирках, метят различными флуоресцентными красителями с разной длиной волны флуоресценции. Затем содержимое всех четырех пробирок соединяют и проводят электрофорез на одной дорожке. Дорожку сканируют в луче лазера и регистрируют положение каждой флуоресцирующей полосы. Все данные вводят в компьютер, который сопоставляет их и выводит на дисплей нуклеотидную последовательность.
ПОЛИМЕРАЗНАЯ ЦЕПНАЯ РЕАКЦИЯ
Полимеразная цепная реакция (ПЦР) – это эффективный способ получения in vitro большого числа копий специфических нуклеотидных последовательностей. Их амплификация – иногда в миллионы раз – осуществляется в ходе трехэтапного циклического процесса. Для ПЦР необходимы:
1) два синтетических олигонуклеотидных праймера (длиной примерно по 20 нуклеотидов), комплементарные участкам ДНК из противоположных цепей, фланкирующим последовательность-мишень; их З'-гидроксильные концы после отжига с ДНК должны быть ориентированы навстречу друг другу;
2) ДНК-мишень длиной от 100 до -35 000 п. н.;
3) термостабильная ДНК-полимераза, которая не теряет своей активности при температуре 95° и выше;
4) четыре дезоксирибонуклеотида.
Типичная ПЦР-амплификация состоит в многократном повторении следующих трех реакций.
1. Денатурация. Первый этап ПЦР состоит в тепловой денатурации образца ДНК выдерживанием его при температуре 95 "С в течение по крайней мере 1 мин. Помимо ДНК, в реакционной смеси содержатся в избытке два праймера, термостабильная ДНК-полимераза Taq, выделенная из бактерий Thermus aquaticus, и четыре дезоксирибонуклеотида.
2. Ренатурация. Температуру смеси медленно понижают до -55 "С, при этом праймеры спариваются с комплементарными последовательностями ДНК.
3. Синтез. Температуру повышают до ~75 "С – величины, оптимальной для ДНК-полимеразы Taq. Начинается синтез комплементарной цепи ДНК, инициируемый 3'-гидроксильной группой праймера (рис. 5.18).
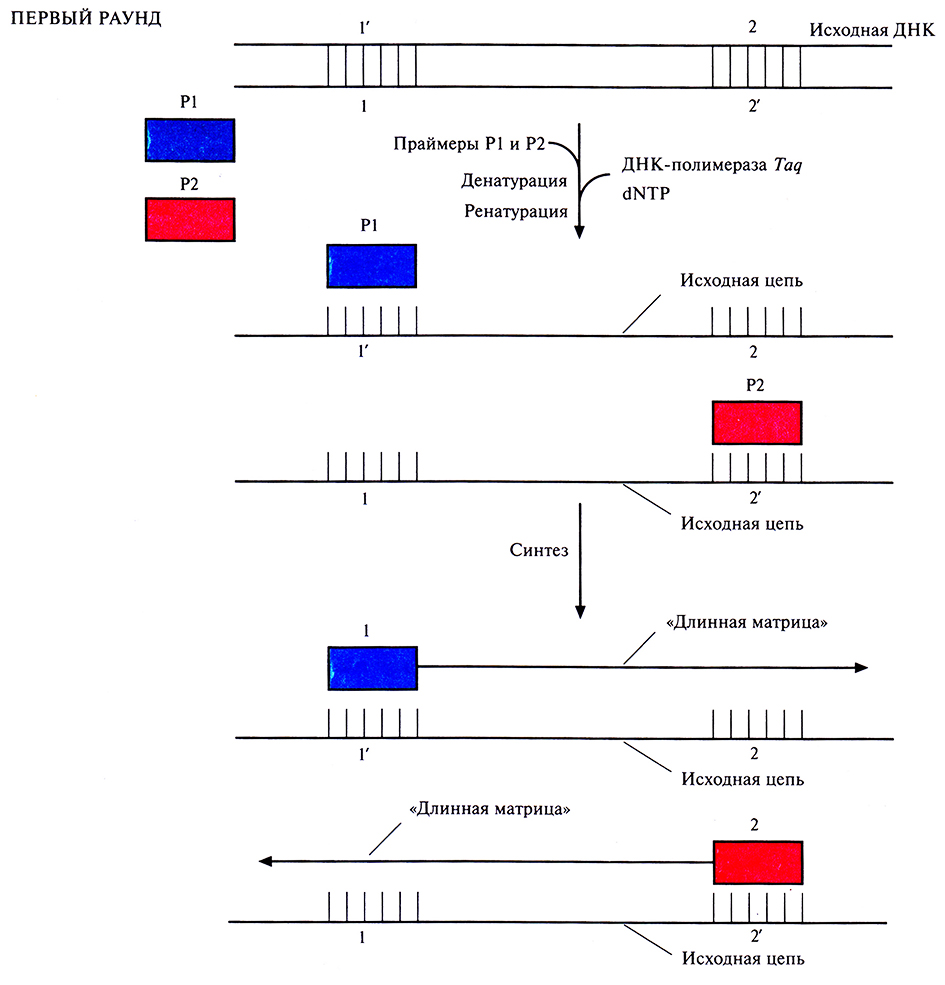
Рис. 5.18. Первый раунд ПЦР. ДНК-мишень фланкирована последовательностями 1'–2 в одной цепи и последовательностями 1–2' – в другой. К образцу ДНК добавляют праймеры (Р1 и Р2), ДНК-полимеразу Taq и четыре дезоксирибонуклеозидтрифосфата (dNTP). Смесь нагревают до 95 °С, инкубируют в течение 1 мин и медленно охлаждают до 55 °С. При этой температуре праймеры, добавленные в избытке, спариваются с разделенными цепями. Повышают температуру до 75 °С. В этих условиях происходит синтез обеих цепей ДНК, начинающийся с З'-гидроксильных концов праймеров. Каждая из синтезированных цепей имеет гораздо большую длину, чем расстояние от З'-гидроксильной группы «ее» праймера до концевого нуклеотида последовательности, комплементарной второму праймеру. Эти цепи служат матрицами во втором раунде ПЦР.
Все реакции проводят в пробирках, погруженных в термостат. Смена температурного режима и его поддержание осуществляются автоматически. Каждый цикл обычно длится 3–5 мин.
Чтобы понять, как именно происходит амплификация определенного сегмента ДНК в ходе ПЦР, нужно четко представлять положение всех праймеров и комплементарных им последовательностей в амплифицируемых цепях в каждом раунде. В первом раунде каждая из новосинтезированных цепей имеет гораздо большую длину, чем расстояние от З'-гидроксильной группы «ее» праймера до концевого нуклеотида последовательности, комплементарной второму праймеру. Такие цепи называют «длинными матрицами», именно на них будет идти дальнейший синтез (рис. 5.18).Во втором раунде двухцепочечную ДНК, состоящую из исходной и новосинтезированной («длинная матрица») цепей, опять подвергают денатурации, а затем отжигают с праймерами. Во время синтеза в этом раунде вновь синтезируются «длинные матрицы», а также некоторое количество цепей с праймером на одном конце и с последовательностью, комплементарной второму праймеру, на другом («короткие матрицы») (рис. 5.19). Во время третьего раунда все гетеродуплексы, образовавшиеся ранее, одновременно подвергаются денатурации и отжигу с праймерами, а затем реплицируются (рис. 5.20). В последующих раундах «коротких матриц» становится все больше, и к 30-му раунду их число уже в 106 раз превышает число исходных цепей или «длинных» матриц (рис. 5.21).
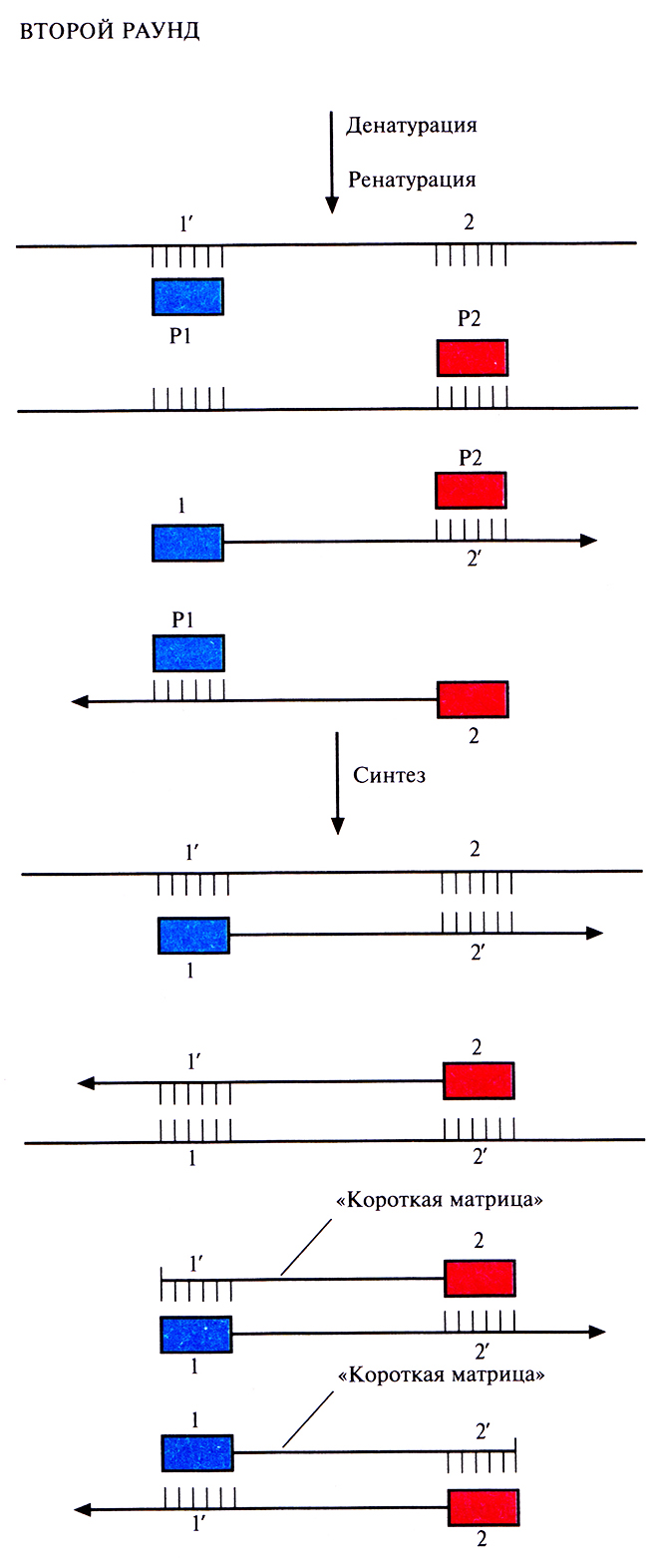
Рис. 5.19. Второй раунд ПЦР. Исходным материалом в этом случае является смесь молекул ДНК, образовавшихся в первом раунде (рис. 5.18). При отжиге праймеры гибридизуются с комплементарными им участками как исходных цепей, так и «длинных матриц», синтезированных в первом раунде. В результате ферментативного синтеза in vitro на исходных цепях синтезируются «длинные матрицы», а на «длинных матрицах» – «короткие». Последние начинаются с одного праймера, а заканчиваются последовательностью, комплементарной второму праймеру.
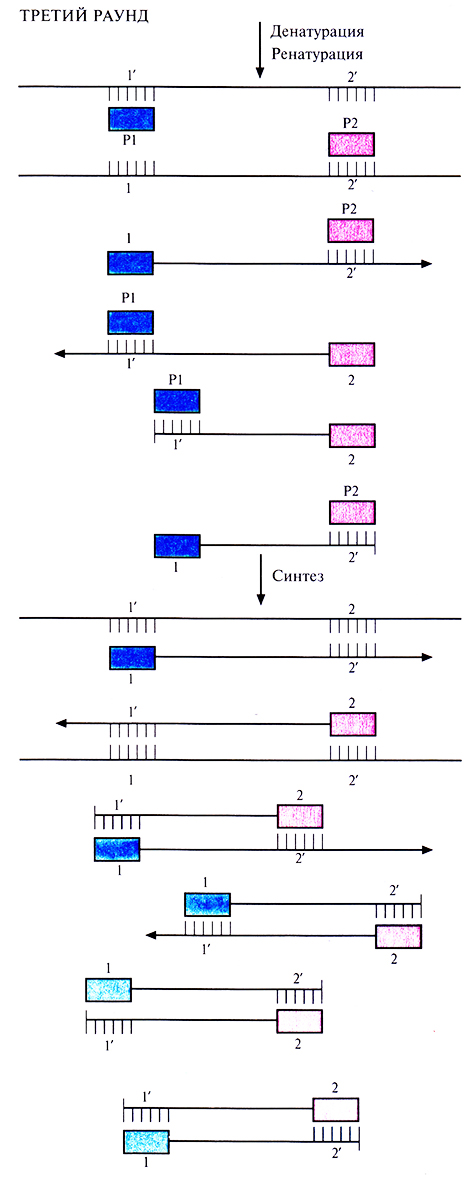
Рис. 5.20. Третий раунд ПЦР. При отжиге праймеры гибридизуются с комплементарными участками исходных цепей, а также «длинных» и «коротких» матриц. При ферментативном синтезе in vitro на исходных цепях синтезируются «длинные» матрицы», а на «длинных» и «коротких» матрицах – только «короткие матрицы».
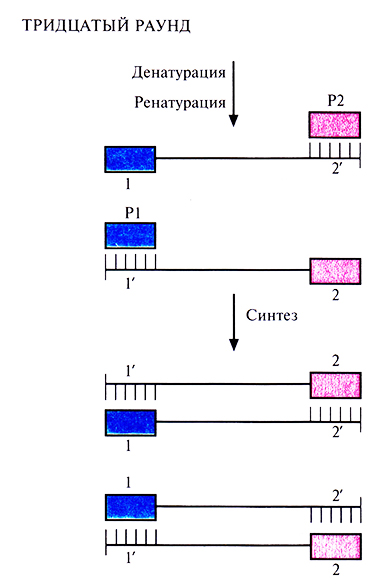
Рис. 5.21. Тридцатый раунд ПЦР. На этом этапе в реакционной смеси содержатся практически одни «короткие матрицы».
Метод ПЦР получил широкое распространение. Разнообразные случаи его применения мы рассмотрим в последующих главах. Здесь упомянем лишь некоторые из них. Один из важнейших – идентификация патогенных микроорганизмов, возбудителей заболеваний человека, животных и растений. С появлением ПЦР отпала необходимость в выделении и очистке ДНК-мишени; для анализа можно использовать очень небольшое количество неочищенного материала. Для синтеза праймеров, специфичных в отношении исключительно ДНК-мишени, нужно знать нуклеотидную последовательность ДНК предполагаемого патогенного микроорганизма. В этом случае в ходе ПЦР будет амплицицироваться только фрагмент ДНК, длина которого равна суммарной длине двух праймеров и фрагмента ДНК между ними.
ПЦР – высокочувствительный метод, поэтому при наличии в исследуемом образце даже ничтожного количества ДНК, случайно попавшей из одной реакционной смеси в другую, могут быть получены ложноположительные результаты. Это заставляет тщательно контролировать все используемые для ПЦР растворы и посуду.
Метод ПЦР применяется также для выявления спонтанных мутаций, внесения специфических мутаций in vitro, сборки полноразмерных генов из синтетических олигонуклеотидов, секвенирования ДНК. Во многих случаях возникает необходимость в клонировании ПЦР-продукта. Однако прямое клонирование с помощью дотирования по тупым концам затруднено, поскольку полимераза Taq присоединяет к 3'-концу синтезируемой цепи лишний адениннуклеотид, что снижает эффективность лигирования. Но если вектор для клонирования обработать рестрицирующей нуклеазой с образованием новых тупых концов и затем проинкубировать с полимеразой Taq в присутствии dTTP, то к обоим 3'-концам фрагментов добавится по одному тимидиннуклеотиду. Взаимной комплементарное™ концевых участков вектора и ПЦР-продукта протяженностью в один-единственный нуклеотид оказывается достаточно для спаривания молекул и их последующего лигирования.
Получение с помощью ПЦР кДНК, отвечающих концам молекул мРНК
С помощью ПЦР можно получать комплементарные ДНК (кДНК), отвечающие 3'- или 5'-концевым участкам специфических информационных РНК (мРНК). Для обозначения этого метода используется сокращение RACE – от англ. rapid amplification of сDNA ends (быстрая амплификация концов кДНК). Обозначения 3' RACE и 5' RACE относятся к амплификации кДНК, отвечающих соответствующим концам мРНК. В обоих случаях для проведения ПЦР-амплификации нужно знать нуклеотидную последовательность кодирующей области мРНК-мишени, чтобы синтезировать генспецифичный праймер (GSP). В случае 3' RACE праймером для синтеза первой цепи кДН К служит oligo(dT) с присоединенным к нему вторым праймером (Р) (рис. 5.22). Oligo(dT) спаривается с poly(A)-хвостом мРНК, и обратная транскриптаза синтезирует цепь, комплементарную мРНК. Вторая цепь кДНК синтезируется на первой при участии GSP, комплементарного кодирующей области данной мРНК, с помощью полимеразы Taq. По завершении нескольких раундов ПЦР, в которых использовались указанные выше праймеры, добавляют вторую пару праймеров, которые связываются по соседству с двумя первыми. Такие тесно расположенные праймеры называются внутренними. Вторая пара праймеров необходима, поскольку без них нельзя амплифицировать полноразмерную молекулу-мишень. Конечным ПЦР-продуктом является кДНК, соответствующая 3'-концу искомой мРНК.
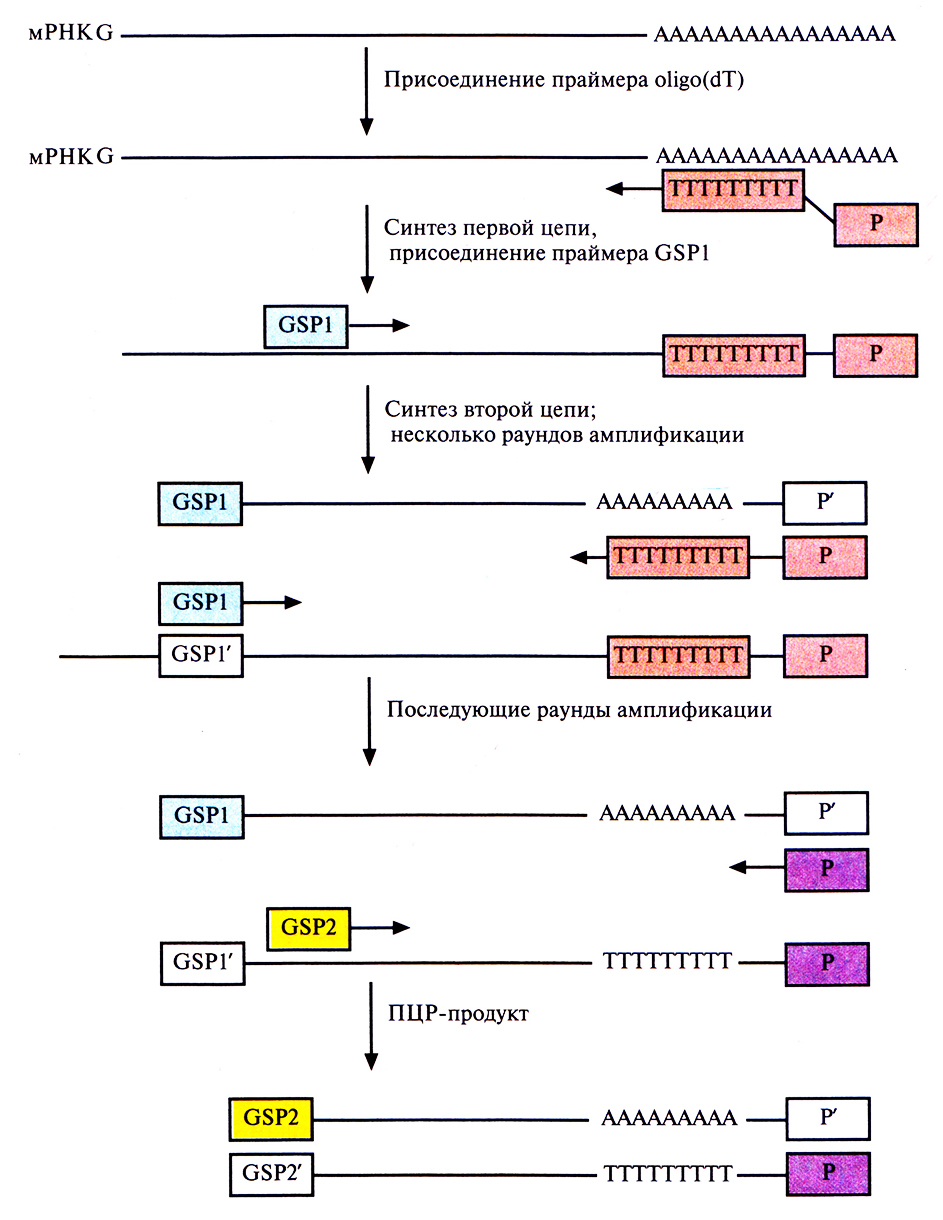
Рис. 5.22. ПЦР-амплификация кДНК, комплементарной 3'-концевой части мРНК. Первая цепь кДНК образуется в результате обратной транскрипции мРНК при участии oligo(dT) в качестве праймера. Вторая цепь синтезируется на первой цепи как на матрице с помощью полимеразы Taq и генспецифичного праймера (GSP1). В последующих раундах ПЦР используются праймеры GSP2 и Р.
В случае 5'RACE праймером для синтеза первой цепи кДНК служит GSP (рис. 5.23). Новосинтезированную цепь обрабатывают концевой дезоксинуклеотидилтрансферазой в присутствии dATP. Этот фермент случайным образом присоединяет дезосирибонуклеотиды к 3'-концу цепи. Поскольку в данном случае в реакционной смеси присутствует только dATP, на этом конце появляется цепочка адениновых остатков – ро1у(А)-хвост. С ним спаривается праймер P-oligo(dT), инициирующий синтез второй цепи. Проводят ограниченное число раундов ПЦР с указанными праймерами, а затем добавляют вторые праймеры и амплифицируют кДНК, отвечающую 5'-концу мРНК.
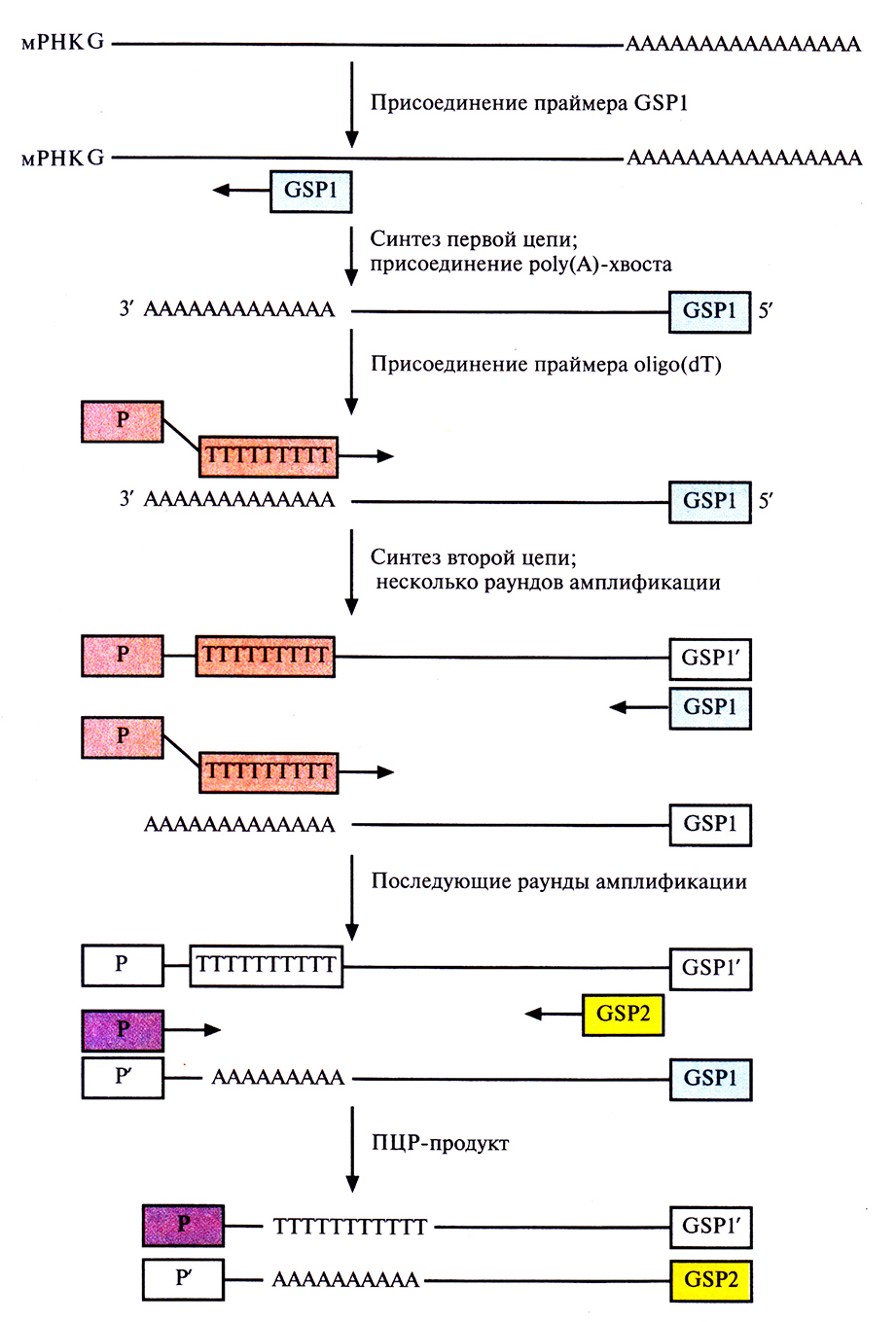
Рис. 5.23. ПЦР-амплификация кДНК, комплементарной 5 концевой части мРНК первой цепи, осуществляемая обратной транскриптазой, инициируется праймером GSP. Затем к этой цепи с помощью концевой дезоксинуклеотидилтрансферазы присоединяется роlу(А)-хвост. Для синтеза второй цепи в качестве праймера используется oligo(dT) Проводят несколько раундов амплификации при участии указанных праймеров, добавляют вторые праймеры (GSP2 и Р) и получают кДНК, отвечающую 5'-концу мРНК.
RACE-метод широко применяется по ряду причин. Обычно бывает очень трудно обнаружить кДНК, соответствующую мРНК, которая присутствует в данной ткани в маленькой концентрации. С помощью RACE-метода можно быстро получить кДНК, отвечающие концевым участкам этой мРНК, и при необходимости использовать их в качестве зондов для скрининга кДНК- и геномных библиотек. Кроме того, поскольку неполноразмерные 3'-концевые фрагменты кДНК значительно преобладают над полноразмерными, 5'RACE может восполнить недостающие 5'-концевые сегменты.
Синтез генов с помощью ПЦР
Получение генов с помощью ПЦР – гораздо более быстрый и экономичный метод, чем тот, который основан на отжиге олигонуклеотидов с перекрывающимися концами, заполнении брешей с помощью ДНК-полимеразы и сшивании разрывов ДНК-лигазой. В одной из методик конструирование гена начинается с отжига двух перекрывающихся олигонуклеотидов (А и В), отвечающих центральной части гена (рис. 5.24). После отжига образуется дуплекс с заглубленными З'-гидроксильными группами, служащими точками инициации синтеза комплементарных цепей при ПЦР. Затем в реакционную смесь добавляют еще два олигонуклеотида, С и D. 3'-конец олигонуклеотида С идентичен 5'-концу олигонуклеотида А, а сам этот олигонуклеотид отвечает участку конструируемого гена, непосредственно примыкающему к его центральной части слева. Аналогично, 3'-конец олигонуклеотида D идентичен 5'-концу олигонуклеотида В и отвечает участку гена, примыкающему к его центральной части справа. После денатурации смеси и отжига образуются дуплексы с протяженными выступающими одноцепочечными сегментами, достраивающимися с 3'-концов В ходе последующих раундов ПЦР образуется двухцепочечный продукт, со стоящий из указанных выше сегментов, расположенных в порядке CABD. Молекула ДНК с заглубленными 5'-концами не достраивается.
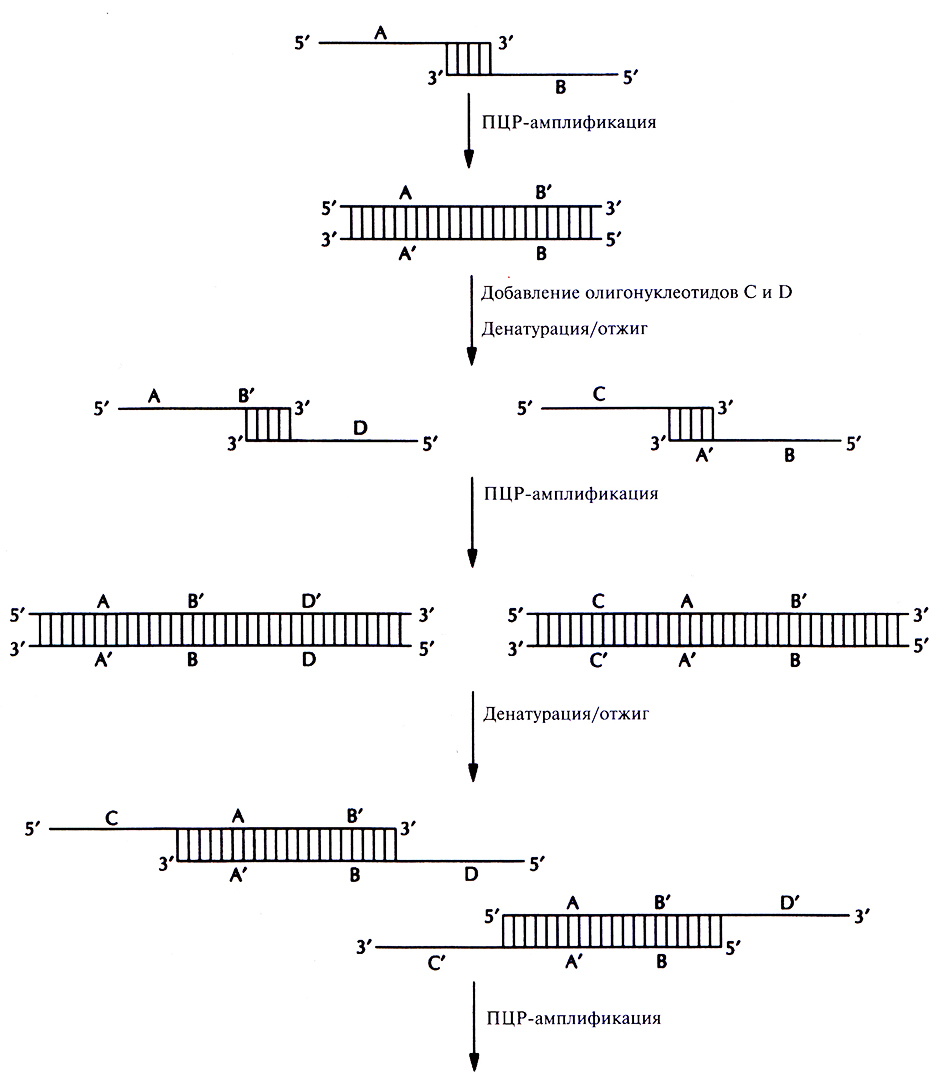
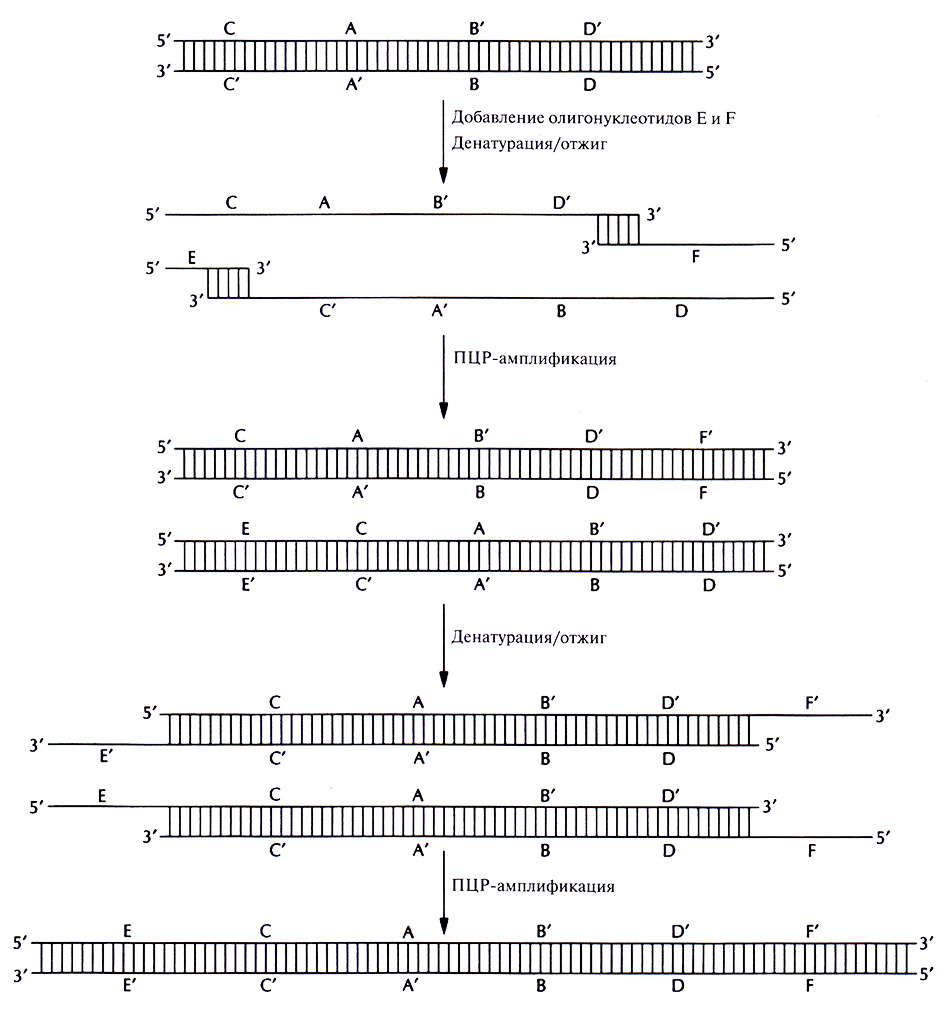
Рис. 5.24. Синтез генов с помощью ПЦР. Перекрывающиеся олигонуклеотиды (А и В) отжигают и достраивают образовавшийся дуплекс с заглубленными З'-гидроксильными концами. Двухцепочечные молекулы денатурируют, добавляют в реакционную смесь вторую пару олигонуклеотидов (С и D), перекрывающихся с продуктами первого раунда ПЦР, и отжигают. Осуществляют второй раунд ПЦР, добавляют следующую пару олигонуклеотидов (Е и F), осуществляют третий раунд ПЦР и т. д. В результате образуется двухцепочечная ДНК, идентичная искомому гену. Одинаковыми буквами со штрихом или без (А' и А, В и В' и т. д.) обозначены комплементарные участки ДНК. Нуклеотидная последовательность каждого олигонуклеотида соответствует таковой определенных сегментов ДНК.
На следующем этапе в реакционную смесь добавляют еще два олигонуклеотида, Е и F. 3'-конец олигонуклеотида Е идентичен 5'-концу олигонуклеотида С, а сам он отвечает участку реконструируемого гена, примыкающему слева к сегменту С. Аналогичными свойствами обладает олигонуклеотид F, если его соотносить с олигонуклеотидом D. После денатурации и ренатурации смеси образовавшиеся дуплексы с выступающими одноцепочечными участками достраиваются с З'-гидроксильных концов. В ходе последующих раундов ПЦР образуется двухцепочечный продукт ECABDF.
Следующие пары олигонуклеотидов – один «достраивающий» ген слева, другой справа – последовательно добавляют в смесь до тех пор, пока не будет синтезирован весь ген. Длина этих олигонуклеотидов обычно бывает равна 50 звеньям. Каждый «блок» ПЦР состоит из двадцати 4-минугных раундов. Для синтеза гена длиной 1000 п. н. нужно 10 «блоков», так что ген можно получить в течение одного дня. При этом, как и в случае синтеза генов другими методами, последнюю пару нуклеотидов (т. е. 5'- и 3'-концы) можно снабдить дополнительными последовательностями, фланкирующими кодирующий участок и облегчающими последующее встраивание гена в вектор.
ЗАКЛЮЧЕНИЕ
К числу наиболее важных для молекулярной биотехнологии методов, помимо клонирования генов, относятся методы химического синтеза ДНК, секвенирование ДНК и полимеразная цепная реакция (ПЦР).
Целью химического синтеза является получение одноцепочечных молекул ДНК in vitro. Успеха здесь можно достичь только при высокой эффективности образования фосфодиэфирных связей. В противном случае по окончании процесса будет получено очень небольшое число молекул нужного размера. Длина синтезированных in vitro молекул обычно составляет 20–30 нуклеотидов и редко превышает 100 нуклеотидов. Для получения двухцепочечных молекул комплементарные цепи синтезируют по отдельности и затем проводят отжиг. Полученную таким способом ДНК используют в качестве зондов для скрининга геномных библиотек; в качестве линкеров и адапторов при клонировании генов; для мутагенеза in vitro; для конструирования генов с целью последующего клонирования.
Очень часто для решения биотехнологических и некоторых других задач бывает необходимо знать полную нуклеотидную последовательность клонированного гена. Для секвенирования используют несколько методов; один из них – дидезокси-метод, разработанный Сангером и др. В его основе лежит остановка синтеза цепи после присоединения к ней дидезоксинуклеотида. У такого нуклеотида отсутствует З'-гидроксильная группа, и дальнейший рост цепи становится невозможным. Для секвенирования в разных пробирках одновременно проводят четыре реакции синтеза ДНК, каждая – в присутствии одного из четырех дидезоксинуклеотидов. Продукты реакций разделяют с помощью гель-электрофореза, проводят радиоавтографию и «считывают» с радиоавтографа нуклеотидную последовательность синтезированного фрагмента ДНК.
Для секвенирования используют также систему на основе фага М13. В ДНК фага встраивают фрагмент ДНК длиной до 500 нуклеотидов, который хотят секвенировать. Эту рекомбинантную ДНК легко получить в одноцепочечной форме и использовать ее в качестве матрицы для секвенирования вставки. Можно использовать также двухцепочечные плазмиды, содержащие клонированную ДНК.
Для определения нуклеотидной последовательности протяженных клонированных сегментов сначала подбирают синтетический олигонуклеотидный праимер, комплементарный участку, соседствующему со вставкой, и с помощью дидезокси-метода секвенируют первые 250–300 нуклеотидов. Затем по результатам секвенирования синтезируют второй праймер и определяют последовательность следующих 250–350 нуклеотидов клонированного участка, и т. д. Этот метод, называемый «праймер-опосредованной прогулкой» (или «блуждающей затравкой»), позволяет секвенировать протяженные фрагменты ДНК без их субклонирования, как в случае системы на основе фага М13.
Метод ПЦР произвел настоящую революцию в биотехнологии. Он позволяет в миллионы раз амплифицировать in vitro нужные сегменты ДНК. Процедура состоит в следующем. Подбирают два праймера, гибридизующиеся с участками ДНК, которые фланкируют искомую последовательность. Денатурируют ДНК, отжигают одноцепочечные молекулы с праймерами, добавленными в избытке, и осуществляют синтез ДНК in vitro. Для облегчения синтеза используют термостабильную ДНК-полимеразу, которая не разрушается при температуре денатурации (95 °С). Затем опять проводят денатурацию, отжиг с праимерами и синтез, и т. д. до примерно 30-го раунда. К этому времени в реакционной смеси преобладают фрагменты, на одном конце которых находится одна праймерная последовательность, а на другом – последовательность, комплементарная второму праймеру. ПЦР можно использовать для выявления патогенных микроорганизмов в том или ином биологическом материале; получения больших количеств специфических фрагментов ДНК с целью клонирования; амплификации 5'- и 3'-концов специфических мРНК; синтеза генов; выявления делеций или вставок в генах, ответственных за то или иное наследственное заболевание.
