Подрекомбинантными понимают ДНК, образованные объединением in vitro двух или более фрагментов ДНК, выделенных из различных биологических источников. Определенные фрагменты ДНК, в том числе и фрагменты, содержащие гены, получают с использованием рестриктаз. Рестриктазы могут образовывать фрагменты как с «тупыми», так и с «липкими» концами. Соединение фрагментов ДНК в единую молекулу производится несколькими методами, зависящими от того, какие концы имеют фрагменты сшиваемых ДНК.
Соединение фрагментов по одноименным «липким» концам
Некоторые рестриктазы, например EcoR I, вносят в цепи ДНК симметричные, расположенные с образованием «ступеньки» разрывы на равных расстояниях от центра сайта узнавания. Эти комплементарные друг другу участки имеют тенденцию к ассоциации за счет спаривания оснований и поэтому их называют комплементарными, или «липкими» концами (рис. 1.4).
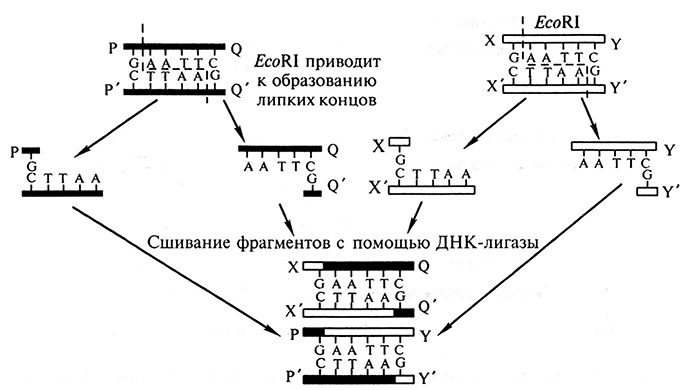
Рис. 1.4. Сшивание фрагментов ДНК с одноименными «липкими» концами
Спаривание оснований происходит только между комплементарными последовательностями, поэтому ААТТ-концы, образующиеся при действии рестриктазы EcoR I, не будут спариваться, например, с AGCT-концами, образуемыми Hind III. Но любые два фрагмента (независимо от их происхождения), образовавшиеся под действием одной и той же рестриктазы, могут «слипаться» за счет образования водородных связей межцу однонитевыми участками комплементарных нуклеотидов (см. рис. 1.4).
Однако после такого спаривания целостность двойной спирали не восстановится, поскольку останется два разрыва в фосфодиэфирном остове. Для их восстановления, т.е. сшивания, или лигирования нитей используют фермент ДНК-лигазу. Таким образом, лигаза завершает образование рекомбинантной молекулы ДНК. Впервые такие эксперименты были выполнены в 1972 г. (П. Берг, США) и было показано, что использование рестриктазы, дающей «липкие» концы, в сочетании с ДНК-лигазой может послужить основой для создания общего метода получения рекомбинантных молекул ДНК. В этой лаборатории впервые были соединены рекомбинантное ДНК вируса SV40 и бактериофага λ.
Соединение фрагментов по «тупым» концам
«Тупые» концы фрагментов ДНК, полученные последействия рестриктаз, производящих прямой разрез внутри сайта рестрикции, также могут быть соединены за счет действия ДНК-лигазы. В этом случае реакция лигирования имеет свои особенности и ее эффективность на порядок ниже, чем при сшивке по «липким» концам. Однако преимущество соединения фрагментов с «тупыми» концами по сравнению с «липкими» в том, что не имеет значения, какие рестриктазы образовывали эти «тупые» концы: фрагменты, образованные в результате действия разноименных рестриктаз, легко соединимы (рис. 1.5).
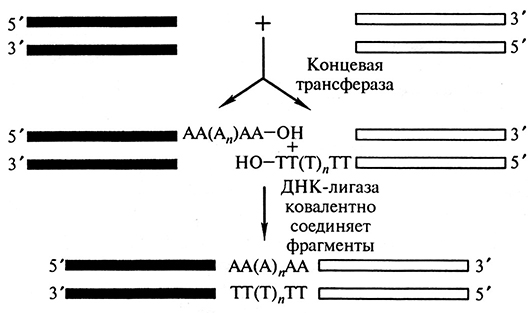
Рис. 1.5. Сшивание фрагментов ДНК с одноименными «тупыми» концами
Соединение фрагментов с разноименными концами
«Липкие» концы можно ферментативным путем присоединить к молекулам ДНК с «тупыми» концами. Для этого «затупляют» «липкие» концы. Это достигается либо отщеплением нуклеотидов «липких» концов с помощью фермента – нуклеазы S1, которая разрушает только одноцепочечную ДНК, либо «липкие» концы «застраивают», т.е. с помощью ДНК-полимеразы I на однонитевых «липких» концах синтезируют вторую нить.
Таким образом, из фрагмента ДНК с «липкими» концами получается фрагмент с «тупыми», и он соединяется с другим фрагментом ДНК с «тупыми» концами ДНК-лигазой.
После того как фрагменты ДНК соединены в пробирке, их надо ввести в живые клетки. Для этого используют специальные молекулы – векторы.
Векторные молекулы. Трансформация
Ключевой операцией в генетической инженерии является введение в клетку и стабильное поддержание генетической информации, содержащейся в рекомбинантных молекулах ДНК. Это достигается при помощи так называемых векторных молекул, или векторов. Дело в том, что при обычном введении ДНК, например в бактериальную клетку, она, как правило, подвергается атаке ферментов, которые гидролизуют ее на составные компоненты – нуклеотиды. В некоторых случаях ДНК «выживает» в клетке, однако в процессе деления клеток она не наследуется и теряется. Для того чтобы рекомбинантная ДНК стала составной частью генетического аппарата клетки, она должна либо встроиться в ее геном (интегрироваться в хромосому) и реплицироваться за ее счет, либо быть способной к автономной репликации.
Векторами называются молекулы ДНК, способные акцептировать чужеродную ДНК и обеспечивающие ее репликацию, экспрессию и/или трансформацию (перенос в другие организмы). Таким образом, вектор позволяет осуществить введение в клетку дополнительной генетической информации. В качестве векторов используют, как правило, плазмиды, бактериофаги, мобильные элементы, вирусы животных. В настоящее время создано большое число векторов, и по профилю использования их можно разделить на несколько типов.
1. Векторы для клонирования используют для увеличения количества (амплификации) фрагмента ДНК, встроенного в такой вектор, посредством репликации. В этом качестве наиболее часто используются бактериальные плазмиды и фаги. Для клонирования больших фрагментов генома используют векторы – искусственные бактериальные и дрожжевые хромосомы (ВАС и YAC).
2. Экспрессионные векторы используют для анализа конкретных последовательностей генов и их белковых продуктов, а также наработки конкретного белка. Существует огромное количество экспрессионных систем, особенно для прокариотических организмов. Есть также векторы для экспрессии генов в клетках дрожжей, растений и млекопитающих. Экспрессионные векторы для эукариотических организмов всегда содержат так называемую экспрессионную кассету, состоящую из промотора, способного работать в данном организме, и сайта полиаденилирования.
3. Векторы для трансформации используют для введения чужеродного фрагмента ДНК в геном реципиента. Обычно такие векторы содержат специфические последовательности, способствующие интеграции в геном.
Современные векторные системы часто бывают полифункциональными, совмещая несколько функций в одном векторе. Первые естественные векторные плазмиды были выделены из бактерий; в последующем большинство векторов было сконструировано при помощи методов генетической инженерии в соответствии с задачами экспериментаторов (экспрессионные векторы, векторы для клонирования, векторы для трансформации).
Часто в составе векторной молекулы бывает маркерный ген, который после проникновения вектора в клетку придает ей фенотип, свидетельствующий о присутствии вектора. Иными словами, вектор должен иметь селективный генетический признак. Часто в качестве селективных используют широко распространенные в природе гены устойчивости к антибиотикам. Белковые продукты этих генов, обычно ферменты, модифицирующие антибиотики и инактивирующие их действие. Например, ген nptll, кодирует фермент неомицинфосфотрансферазу, инактивирующий антибиотик канамицин. В присутствии гена nptll бактериальная клетка приобретает устойчивость к канамицину и на среде с этим антибиотиком образует клон или колонию клеток. Обычные же клетки, не содержащие ген nptll, на данной среде погибают. Таким образом, присутствие маркерного гена nptll в векторной конструкции позволяет обнаружить те клетки, в которых присутствует вектор, и отобрать их.
Трансформация клеток Е. coli векторными конструкциями
Полученные векторные конструкции, содержащие чужеродный фрагмент ДНК, используют для трансформации бактериальных клеток специальных штаммов Е. coli. Трансформация бактерий плазмидами векторов основана на способности клеток акцептировать внутрь себя молекулы ДНК (компетентность клеток). Трансформацию клеток Е. coli обычно проводят одним из двух методов: с помощью кальциевого шока или электропорацией. В обоих случаях бактериальная мембрана становится более проницаема для молекул ДНК, последние входят в протоплазму бактериальной клетки.
В целом к векторной молекуле предъявляются следующие основные требования:
1) вектор должен содержать уникальные сайты рестрикции для нескольких рестриктаз, что делает возможным встроить в него фрагмент чужеродной ДНК;
2) вектор должен обладать определенной емкостью и не абортировать встроенный фрагмент;
3) вектор должен реплицироваться в определенных клетках за счет имеющейся последовательности точки начала репликации (ориджина);
4) вектор должен содержать последовательность маркерного гена, облегчающего селекцию клеток, несущих векторную конструкцию.
Использование бактериальных плазмид в качестве векторов для клонирования
Клетки бактерий содержат хромосомную ДНК. Однако помимо хромосом бактерии содержат большое число небольших (1–25 т. н. п.) кольцевых молекул ДНК. Такие кольцевые молекулы называют плазмидами. Некоторые плазмиды имеют в своем составе гены устойчивости к антибиотикам, представленные большим числом копий. Высокая копийность плазм ид обеспечивает клетке синтез большого количества ферментов, биохимически нейтрализующих антибиотики, что и обеспечивает устойчивость бактериальной клетки к последним.
Как было сказано выше, в присутствии ионов кальция плазмиды легко поглощаются бактериями, которые их не содержали. Однако бактериальная клетка обычно может содержать в своем составе плазмиды только одного типа.
Число копий плазмиды в клетке может существенно варьировать. Это зависит от генетических особенностей как клетки, так и плазмиды. Некоторые плазмиды могут размножаться до тех пор, пока их число не достигнет 10–200 копий на клетку. Другие типы плазмид реплицируются с той же скоростью, что и бактериальная хромосома. Такие плазмиды содержатся в клетке в количестве одной или нескольких копий. Естественно, для целей клонирования используют векторы на основе плазмид первого типа.
Впервые плазмида в качестве вектора была использована в 1973 г. в лаборатории П. Берга. Эксперименты проводились с небольшой (~9 т. н. п.) плазмидой Е. colipSC101, несущей ген устойчивости к антибиотику тетрациклину. Она содержала только один сайт рестрикции для EcoR I. Под действием рестриктазы кольцевая плазмида превращалась в линейную молекулу с «липкими» концами. Такую ДНК плазмиды pSC101 смешивали с EcoR I-фрагментом чужеродной для кишечной палочки ДНК (ДНК золотистого стафилококка). С помощью ДНК-лигазы фрагмент чужеродной ДНК и плазмидырБСКИ соединяли в единую рекомбинантную молекулу. Затем такую рекомбинантную плазмиду добавляли к компетентным клеткам Е. coli, плазмида входила внутрь бактериальной клетки. Клетки с рекомбинантной плазмидой отбирались на селективной среде с тетрациклином (рис. 1.6). Этот исторический опыт положил начало генетической инженерии. Стало ясно, что можно будет проводить дальнейшие эксперименты с встраиванием различных фрагментов чужеродных про- и эукариотических ДНК и получать клетки с новыми свойствами.
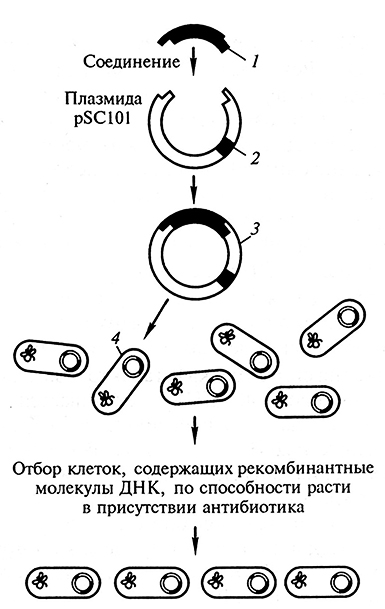
Рис. 1.6. Схема эксперимента по клонированию фрагментов ДНК с помощью плазмид:
1 – встраиваемая гетерологичная ДНК; 2– маркер устойчивости к антибиотику;
3 – рекомбинантная молекула ДНК; 4 – введение в бактериальные клетки
В настоящее время на основе природных векторов сконструированы более удобные в использовании векторы. Наибольшее распространение приобрели векторы типа pUC и их производные (pBluescript, pGEM), которые используются как для клонирования, так и для экспрессии. Векторы pUC небольшие (~ 2,7 т. н. п.), мультикопийные плазмиды, содержат последовательность точки начала репликации (ori-сайт), что позволяет им независимо реплицироваться в бактериальной клетке. Для облегчения клонирования в векторе pUC имеется искусственно синтезированная последовательность полилинкера, представляющая собой уникальные сайты рестрикции для наиболее часто использующихся рестриктаз (рис. 1.7). Полилинкер встроен в ген lacZ, который является удобным маркером на присутствие в векторе чужеродной ДНК. Ген lacZ кодирует белок β-галактозидазу, способный расщеплять субстрат X-gal (производное β-галактозида). При этом цвет бактериальных клеток меняется с белого на ярко-голубой. Небольшая последовательность полилинкра (100–120 н. п.) не влияет на синтез белка β-галактозидазы. При клонировании фрагмент чужеродной ДНК встраивается в полилинкер pUC-вектора, что нарушает экспрессию гена lacZ, клетки, содержащие вектор со вставкой ДНК, не будут расщеплять субстрат X-gal, и колонии таких бактериальных клеток останутся белыми, в отличие от голубых колоний клеток, несущих пустую векторную плазмиду без вставки. Наличие такой бело-голубой селекции pUC-векторов и их производных значительно облегчает процесс клонирования, так как встраивание чужеродной ДНК в векторную плазмиду довольно редкое событие – только одна из 10–30 полученных после лигирования молекул будет рекомбинантной, т. е. нести в своем составе чужеродный фрагмент, и использование бело-голубой селекции позволяет определить только те клоны бактерий, которые содержат рекомбинантную ДНК.
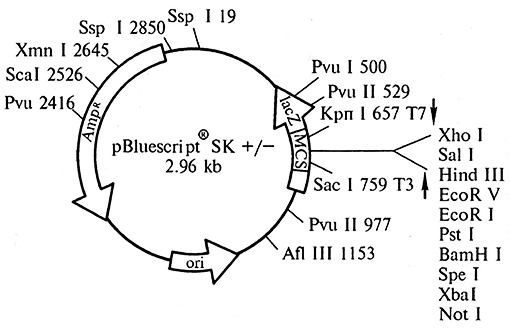
Рис. 1.7. Схема вектора для клонирования pBluescript SK
(MCS – последовательность полилинкера; AmpR – ген устойчивости к ампицилину (селективный маркер)
PUC-векторы несут специфические экспресснойные кассеты, состоящие из регуляторных последовательностей, обеспечивающих транскрипцию клонируемых генов.
Фаговые векторы. Космиды. ВАС- и YAC-векторы
Векторы на основе бактериальных плазмид широко используются для клонирования, но у них есть один важный недостаток – небольшая емкость. В таких векторах можно клонировать фрагменты в среднем не более 7–8 т. н. п., а последовательности эукариотических генов гораздо длиннее (в среднем 10–25 т. н. п). Кроме того, для изучения регуляторных последовательностей, находящихся за пределами кодирующей части генов, необходимо клонировать еще более протяженные области генома. Оказалось, что для клонирования таких фрагментов плазмидные векторы не подходят. Дело в том, что плазмиды, содержащие большие вставки хромосомной ДНК (более 8 т. н. п.), нестабильны и при репликации постепенно уменьшаются в размере в результате делеций нуклеотидов чужеродной ДНК.
На основе бактериофага X были сконструированы векторы, в составе которых фрагменты чужеродной ДНК длиной до 22 т. н. п. весьма стабильны. При создании таких векторов для клонирования на основе фага λ учитывалось то обстоятельство, что вся центральная часть молекулы ДНК фага (∼19 т. н. п.) не нужна для репликации фага в Е. coli. Эту область с помощью рестриктазы вырезают из генома фага так, что правый и левый концевые фрагменты (так называемые правое и левое плечо фага), необходимые для репликации, остаются неизменными. Плечи фага отделяют от остальных фрагментов и используют в качестве векторов для клонирования, содержащих на месте вырезанной фаговой ДНК вставку чужеродной ДНК размером около 9–21 т. н. п. Размножаются в бактериях только те фаги, которые содержат оба конца фаговой хромосомы и вставку чужеродной ДНК (рис. 1.8), при этом длина полученной рекомбинантной ДНК фага не должна быть меньше 30 т. н. п. и больше 52 т. н. п.
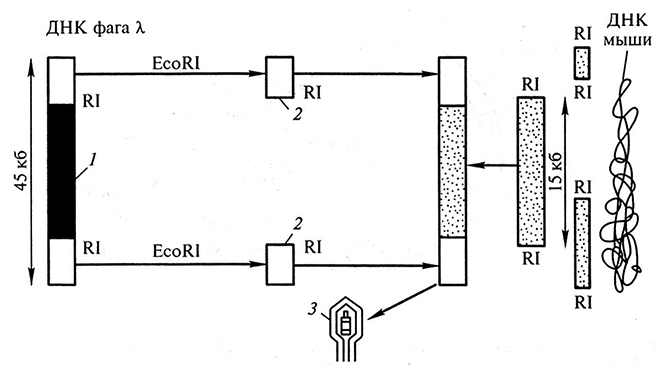
Рис. 1.8. Клонирование ДНК в векторе на основе бактериофага λ:
1 – фрагмент ДНК фага, не нужный для репликации;
2 – фрагменты ДНК фага, содержащие гены, ответственные за репликацию
в бактериальных клетках и дальнейшую упаковку фага;
3 – бактериофаг, содержащий фрагмент чужеродной ДНК
Для выделения и исследования более длинных генов или группы соседних генов с прилежащими к ним последовательностями необходимо клонировать фрагменты ДНК еще большей длины (30–45 т. н. п.). Для клонирования таких фрагментов были сконструированы специальные векторы с большей емкостью – космиды, представляющие собой гибридную молекулу, содержащую специальный cos-участок генома фага, за счет чего они могут упаковываться в головку фага λ, и специальные последовательности, позволяющие им реплицироваться по плазмидному типу. Размер космиды довольно мал по сравнению с фаговым вектором – всего 5 т. н. п. и, следовательно, в космиду можно вставить чужеродную ДНК значительных размеров (30–45 т. н. п.). Фаговые головки, содержащие такую рекомбинантнуюДНК, не могут размножаться как фаги. Ими трансформируют клетки Е. coli. Гибридная молекула, содержащая эукариотическую ДНК, обрамленную cos-еайтами, размножается в Е. coli как плазмида, и каждая фаговая частица вызывает образование колонии индивидуального бактериального трансформанта.
Клонирование фрагментов ДНК от 100 т. н. п. и более осуществляют в специально сконструированных векторах ВАС и YAC.
ВАС-векторы получены на основе F-плазмид бактерий и содержат гены, ответственные за репликацию и копийность этих плазмид в бактериальных клетках, и ряд дополнительных последовательностей для облегчения клонирования. Емкость ВАС-векторов при собственном небольшом размере (~ 7 т. н. п.) огромная – 100–300 т. н. п. Это позволяет во много раз уменьшить число клонов при получении геномных библиотек (см. ниже).
YAC-векторы, которые также используются для клонирования больших фрагментов ДНК, представляют собой искусственную дрожжевую минихромосому. YAC-вектор содержит центромеру, теломеры и точку начала репликации. В такой вектор можно встроить фрагменты чужеродной ДНК размером более 100 т. н. п., и такая минихромосома, введенная в клетки дрожжей, будет реплицироваться и вести себя аналогично другим дрожжевым хромосомам при митотическом делении.
Геномная библиотека (банк генов)
Представляет собой клонированный в составе векторов полный набор последовательностей ДНК данного организма. Фрагментация целого генома на отдельные участки значительно облегчает все генно-инженерные манипуляции и позволяет анализировать отдельные последовательности, проводить сравнительный анализ различных геномов по определенным участкам и, главное, выделять и работать с индивидуальными генами.
Фрагменты геномной ДНК получают гидролизом высокополимерной хромосомной ДНК при помощи рестриктаз. Рестриктазы для гидролиза ДНК подбирают таким образом, чтобы полученные фрагменты перекрывались и их размер приблизительно соответствовал емкости вектора. Полученные фрагменты лигируют с плечами фага и упаковывают в уже готовые головки фаговых частиц. Таким образом, получается геномная библиотека. Библиотеку хранят в виде фагового банка под хлороформом при – 70 °С. Таким способом библиотека может храниться десятки лет. Размножение полученных геномных клонов (амплификацию библиотеки), а также поиск нужного клона проводят, заражая фаговой библиотекой бактериальные клетки и затем высевая их на чашки Петри с агаризованной средой. Полный гаплоидный набор хромосом клетки млекопитающих содержит около 3 • 109 пар оснований. При емкости вектора 15 т. н. п. библиотека может содержать около 1 млн фаговых частиц. Проверка миллиона фаговых бляшек позволяет проверить весь геном на присутствие необходимого гена. Так можно идентифицировать именно те фаговые частицы, которые несут последовательность исследуемого гена, размножить ДНК только этой последовательности и проводить с ней дальнейшие манипуляции.
1.5. ИДЕНТИФИКАЦИЯ И ВЫДЕЛЕНИЕ
ПОСЛЕДОВАТЕЛЬНОСТЕЙ ГЕНОВ
Выделение генов – один из главных этапов в генетической инженерии. Успех на этой стадии создания рекомбинантных ДНК зависит прежде всего оттого, насколько изучен данный ген и его положение в геноме донора; от разработки методов выделения мРНКданного гена и методов обнаружения функциональной активности продукта данного гена; от существования объектов, в которых данный ген находится в больших количествах (амплифицирован) или в которых он особенно активен, что позволяет выделить достаточные количества его мРНК, которые можно в дальнейшем использовать для получения ДНК этого гена путем обратной транскрипции.
В принципе существуют два основных пути получения гена: либо ген искусственно синтезируют, либо из клонотеки отбирают ту рекомбинантную ДНК, которая содержит нужный ген.
Синтез комплементарной ДНК (кДНК)
Осуществление первого пути стало возможным вследствие открытия фермента – обратной транскриптазы илиревертазы. Этот фермент был выделен из некоторых РНК-содержащих онкогенных вирусов. Фермент представляет собой специфическую ДНК-полимеразу и осуществляет РНК-направляемый синтез ДНК, используя РНК как матрицу. В результате синтезируется комплементарная цепь ДНК. Отсюда и название фермента – он катализирует реакцию, обратную первому этапу экспрессии гена – транскрипции.
Предположим, что нам удалось выделить гомогенный препарат мРНК определенного гена. В некоторых случаях, когда эта мРНК составляет существенную часть тотальной мРНК, это удается. Практически все эукариотические мРНК содержат на своем З'-конце последовательность, состоящую из остатков аденина, – поли(А)-последовательность, которая присоединяется к мРНК в результате процессинга.
Для начала реакции синтеза ДНК ДНК-ревертазе, как и всем ДНК-полимеразам, необходима затравка в виде небольшого двуцепочечного отрезка. Если добавить в реакцию короткие одноцепочечные олигонуклеотиды, состоящие из 18–20 тиминовых остатков (поли дТ), то они будут гибридизоваться (образовывать по принципу комплементарности двуцепочечные ДНК–РНК-фрагменты) с поли(А)-последовательностью мРНК – и такой дуплекс будет служить затравкой для начала ферментативной реакции (рис. 1.9). В результате образуется гибридная РНК–ДНК-молекула, причем на конце у нее фермент будет синтезировать короткий отрезок двуцепочечной ДНК – шпильку. Шпилька может служить затравкой для синтеза второй комплементарной цепи ДНК, который осуществляет уже фермент ДНК-полимераза I (рис. 1.9). Цепь мРНК гидролизуют РНКазой. С помощью эндонуклеазы S1, которая специфически гидроролизует одноцепочечные участки ДНК, последовательность шпильки можно расщепить. В результате получится двуцепочечная молекула ДНК, одна из нитей которой комплементарна мРНК. Такую ДНК называют кДНК, и она соответствует структурному гену, с которого транскрибировалась исходная молекула мРНК.
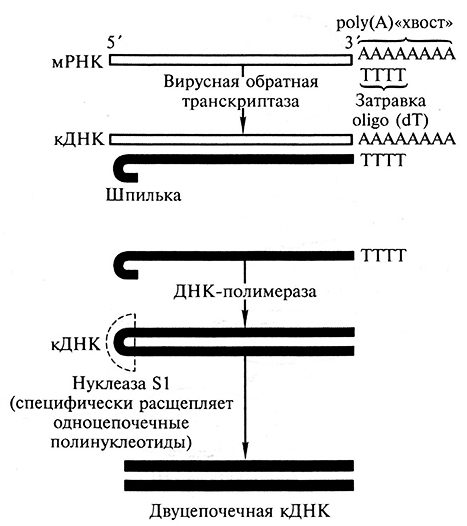
Рис. 1.9. Схема синтеза двуцепочечной кДНК на мРНК
К полученной кДНК присоединяют «липкие» концы и встраивают в плазмиду, например pUC19. Рекомбинантную ДНК вводят в Е. coli, где кДНК-фрагмент размножается. Подобная схема была использована для получения генов, кодирующих инсулин, гормон роста, интерферон, альбумин, иммуноглобулины и ряд других белков, производство которых уже налажено в промышленных масштабах.
Для того чтобы убедиться, что получен именно тот ген, который кодирует данный белок, существует несколько методов. Если известна последовательность аминокислот белка, кодируемого геном, то, определив нуклеотидную последовательность клонированной ДНК (см. рис. 1.3), можно убедиться, что она кодирует именно этот белок.
Иногда применяют метод ДНК – РНК-гибридизации. Это возможно в том случае, если выделена индивидуальная мРНК (рис. 1.10). Молекулы кДНК подвергают денатурации (при этом нити ДНК расходятся) и смешивают с избытком радиоактивно меченой мРНК. В условиях ренатурации (процесс, при котором происходит восстановление двойной спирали, причем последняя может образоваться как из двух цепей ДНК, так и из цепи ДНК и комплементарной ей цепи РНК) будет происходить образование гибридных ДНК – РНК-молекул. Используя эндонуклеазу S1, можно убедиться в их полной комплементарное. Если кДНК не полностью комплементарна РНК, то в гибридной молекуле будут одноцепочечные неспаренные участки, которые могут быть гидролизованы эндонуклеазой S1.
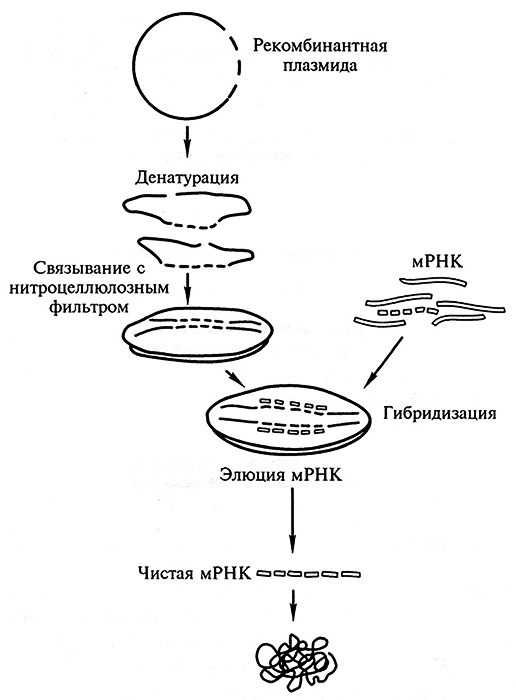
Рис. 1.10. Идентификация плазмид, содержащих в виде вставок специфические кДНК,
с помощью ДНК – РНК-гибридизации на фильтрах
Если кДНК в составе векторной молекулы способна к экспрессии, происходит синтез мРНК, а затем с мРНК – синтез белка, то ген можно идентифицировать по его продукту, например при помощи специфического иммунохимического метода обнаружения белка. Таким образом, используя один из этих методов или их комбинацию, можно доказать, что получен именно нужный ген.
Создание библиотеки кДНК
Мы рассмотрели случай, когда удалось выделить индивидуальную мРНК. Это возможно, если ген избирательно активен в определенных клетках или тканях. В этом случае реакцию обратной транскрипции проводят со смешанной популяцией мРНК и в результате получают кДНК, представляющую смесь обратных транскриптов со всехтипов мРНК. Далее обычно прибегают к методу создания банка кДНК. Для этого молекулы кДНК сшивают с векторными молекулами и трансформируют ими бактериальные клетки. Причем эксперимент проводят таким образом, что каждая бактериальная клетка получает только одну рекомбинантную плазмиду и, следовательно, будет содержать только один вид кДНК. Совокупность векторных плазмид, каждая из которых несет в своем составе кДНК, называют библиотекой кДНК. Таким образом, библиотеки кДНК, в отличие от геномных библиотек, представляют не все последовательности генома и даже не все гены, а только ту их часть, которая экспрессируется в этом организме в этот момент времени. При этом необходимо помнить, что в кДНК представлена только структурная часть гена без промоторных и интронных участков.
Выделяя мРНК только из листьев или только из корней, можно получить специфические кДНК листовой или корневой ткани, которые будут соответствовать только тем генам, которые экспрессируются в определенных тканях: листьях или корнях соответственно. Таким образом, помимо библиотеки кДНК растений или животных можно получить библиотеки кДНК отдельных органов и тканей (органоили тканеспецифичные библиотеки кДНК) или библиотеки кДНК, характерные для различных стадий онтогенеза. Получение и анализ таких библиотек позволяет идентифицировать тканеспецифичные гены и изучать процессы дифференцировки клеток и тканей, ткане- и органоспецифического метаболизма, определять гены, участвующие в этих процессах.
Идентификация нужного гена из клонотеки. В этом случае задача исследователя сводится к поиску среди миллионов клонов тех, которые содержатфрагментДНКс интересующим его геном. В настоящее время для решения такой задачи применяются молекулярные зонды (пробы). Зонд представляет собой меченую молекулу нуклеиновой кислоты, гомологичную последовательности гена.
Индивидуальная радиоактивно меченая мРНК служит хорошим зондом для выявления бактериальных колоний, несущих искомый ген. Однако индивидуальную мРНК не всегда удается выделить. Иногда продукт (а значит, и мРНК) искомого гена присутствует в клетке в очень малом количестве, или мРНК очень быстро деградирует, так что получить индивидуальную РНК не удается. При наличии стабильного белка можно выделить его небольшое количество и определить аминокислотную последовательность некоторой его части (обычно достаточно знания участка белковой молекулы длиной 5–10 аминокислотных остатков). По известной аминокислотной последовательности можно установить возможные последовательности нуклеотидов (их будет несколько из-за вырожденности генетического кода) в том участке мРНК, который кодирует данную аминокислотную последовательность. Затем из отдельных радиоактивно меченых нуклеотидов химически синтезируют олигонуклеотиды длиной не менее 30 нуклеотидных пар, один из которых полностью комплементарен участку искомого гена. Такие нуклеотиды и являются пробами/зондами для поисков нужных клонов, соответствующих кДНК, а затем и клонов в геномных библиотеках.
Скрининг библиотек. Послетого как радиоактивный зонд получен, просматривают геномную библиотеку или библиотеку кДНК для идентификации бактериальных колоний, содержащих плазмиду с последовательностью, комплементарной зонду и, следовательно, соответствующей анализируемому белку.
На чашку Петри с бактериальными колониями накладывают лист специального нейлонового фильтра; на него переходит часть клеток бактерий каждой колонии, тогда как другая часть остается на чашке. В результате получается реплика: на чашке и на фильтре колонии распределены одинаково. ДНК прочно связывается (иммобилизуется) с нейлоновым фильтром. Для лизиса бактерий и денатурации ДНК клонов нейлоновый фильтр обрабатывают щелочью. Затем фильтр помещают в раствор, содержащий меченый зонд (кДНК или мРНК, или синтетический олигонуклеотид). При гибридизации зонд связывается с теми колониями, которые содержат последовательности, комплементарные меченому зонду; фильтр отмывают от несвязавшихся молекул зонда и проводят радиоавтографию, которая выявляет колонии, содержащие метку. Эти колонии идентифицируют на чашке Петри, отбирают и размножают. Таким образом, из огромного числа геномных библиотек или клонов кДНК отбирается только тот, который несёт последовательность анализируемого гена, и в дальнейшем все манипуляции проводятся не со всем геномом, а только с одним геном (фрагментом ДНК).
Анализ ДНК методом блот-гибридизации
Используется не только при скрининге кДНК и геномных библиотек, но и для анализа геномной ДНК. При помощи этого метода можно определить присутствие определенной последовательности ДНК в геноме (например, присутствие чужеродного гена в геноме трансгенных растений, копийность гена, анализировать изменения в нуклеотидной последовательности гена и т. д.). Анализ ДНК методом блот-гибридизации основан на идентификации определенных фрагментов ДНК путем их гибридизации со специфическими мечеными зондами. Он состоит из следующих этапов:
1) рестрикция ДНК;
2) перенос рестрицированных фрагментов из геля на нейлоновый фильтр и их иммобилизация;
3) гибридизация с меченым зондом.
Высокомолекулярную хромосомную ДНК расщепляют одной или несколькими рестриктазами. Образовавшиеся фрагменты разделяют методом электрофореза в агарозном геле, и на предварительно денатурированный (0,4 М NaOH) гель помещают лист нейлонового фильтра. Фильтр покрывают слоями фильтровальной бумаги; под действием капиллярных сил ДНК-фрагменты перпендикулярно переносятся на фильтр и связываются с ним (иммобилизуются). Такой перенос называется блоттинг (от англ. blot – промокать). При этом на фильтре получается как бы реплика с геля. Затем фильтр помещают в раствор с радиоактивно меченым одноцепочечным зондом и гибридизируют с закрепленными на фильтре фрагментами хромосомной ДНК. Зонд гибридизуется только с теми фрагментами, которые содержат гомологичную ему последовательность ДНК. Фрагменты, с которыми связывалась метка, выявляют радиоавтографией. На рис. 1.11 представлена схема проведения блот-гибридизации по Саузерну (Southern blotting).
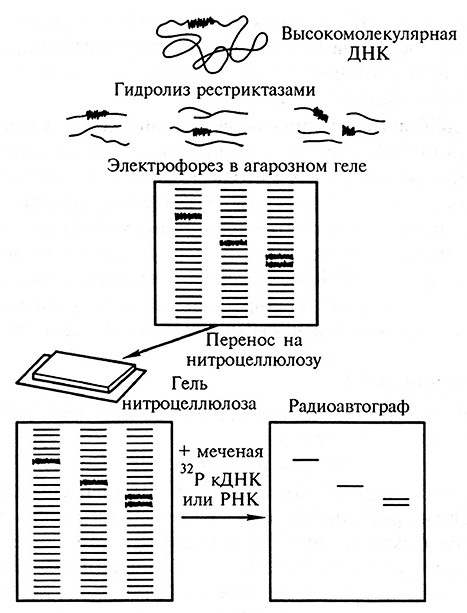
Рис. 1.11. Принцип блот-гибридизации по Саузерну
По полученным на радиоавтографе полосам судят о присутствии анализируемого фрагмента в геноме, изменениях в этой последовательности (делеции, инсерции), а по интенсивности полос можно определить число копий гена в геноме. Таким образом, описанный метод активно используется как для анализа отдельных генов, так и целых геномов.
Аналогичным образом можно им мобилизовывать и анализировать мРНК (Nothern blotting).
Итак, методы в том или ином сочетании позволяют получить многие гены, продукты которых – белки – известны и могут быть выделены хотя бы в малом количестве. Эти гены в дальнейшем могут стать объектом генно-инженерных манипуляций, задача которых получить их экспрессию в новом генетическом окружении.
