Состояние здоровья человека зависит от многих факторов: его образа жизни, биологии, условий окружающей среды, качества системы здравоохранения и т.д. За последние десятилетия достигнуты значительные успехи в диагностике, лечении и профилактике инфекционных болезней, поэтому более очевидным стало влияние генетических факторов, особенно в развитых странах. Например, в Канаде, согласно статистическим данным, у 5% населения в возрасте до 25 лет обнаруживаются наследственные дефекты, приводящие к инвалидности, а у более чем 50% в течение жизни развивается заболевание, имеющее в той или иной степени наследственную природу. В настоящее время более половины случаев обращения в детские лечебные учреждения связаны с генетическими заболеваниями.
Наследственными являются свыше 1000 болезней человека. Большинство из них очень редки (~10-5), но некоторые встречаются относительно часто (~10-4). Многие наследственные заболевания человека обусловливаются мутациями в единственном гене, однако ряд сложных патологий, например рак, определяется мутациями в нескольких генах. В том случае, когда имеется полное, точное и последовательное описание симптомов заболевания (фенотипа), определяемого единственным геном, генетическую природу заболевания можно установить исходя из типа его наследования в семьях, представленных несколькими поколениями. Существуют четыре основных типа наследования: аутосомно-доминантный (рис. 20.1), аутосомно-рецессивный (рис. 20.2), X-сцепленный доминантный (рис. 20.3) и Х-сцепленный рецессивный (рис. 20.4). Термин «аутосомный» относится к 22 парам неполовых хромосом (аутосом) человека, а термин «Х-сцепленный» указывает на локализацию гена на Х-хромосоме. Доминантным называют такое состояние, когда для проявления заболевания достаточно присутствия одного мутантного аллеля данного гена, а в случае рецессивного заболевания дефектными должны быть оба аллеля. У мужчин в ядре присутствует одна Х-хромосома, поэтому большинство Х-сцепленных генов независимо от того, являются они доминантными или рецессивными, приводят к проявлению заболевания.
Анализ родословных чрезвычайно полезен для установления типа наследования специфического состояния, однако не дает никакой информации об ассоциированном с данным заболеванием гене, о биологической основе нарушения или – в случае аутосомного заболевания – о хромосомной локализации гена. Более того, не всегда можно определить, является ли заболевание наследственным. Во-первых, не у всех лиц, несущих дефектный ген, проявляются симптомы заболевания (неполная пенетрантность). Во-вторых, симптомы (фенотип) могут варьировать от слабых до ярко выраженных (варьирующая экспрессивность). В-третьих, один и тот же фенотип может обусловливаться дефектами в совершенно разных генах (генетическая гетерогенность). В-четвертых, в некоторых случаях альтернативные формы (аллели) одного гена могут приводить к разным фенотипам. В-пятых, из-за небольшого размера семей со случаями исследуемого заболевания приходится собирать данные о большом числе родословных, чтобы сделать вывод о природе этого заболевания.
Успех в установлении корреляции между нормальным или патологическим фенотипом, с одной стороны, и соответствующим ему генотипом, с другой, в значительной степени зависит от того, удается ли идентифицировать и изолировать (клонировать) конкретный ген. Зная нуклеотидную последовательность гена, можно определить, какую функцию выполняет его продукт в норме, как нарушается эта функция в результате мутации, в какой степени различные мутации в разных экзонах ответственны за проявление заболевания. Если ген клонирован, можно поставить такие эксперименты, которые позволяли бы установить функции генного продукта, его взаимодействие с другими веществами и характер нарушения нормальных процессов, вызываемого продуктом мутантного гена. Кроме того, основываясь на различиях в нуклеотидных последовательностях нормального и мутантного генов, можно разработать диагностические тесты для выявления специфических мутаций. Чем полнее будут наши знания о функциях гена, ответственного за то или иное заболевание, тем более эффективную схему лечения мы сможем предложить.
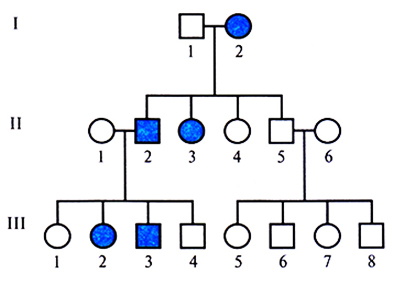
Рис. 20.1. Аутосомно-доминантный тип наследования. Квадратиками изображены мужчины, кружками – женщины; закрашенные символы – больные члены семьи, не закрашенные – здоровые. Горизонтальная линия, соединяющая квадратик и кружок, означает, что данные мужчина и женщина являются супругами. Вертикальные линии ведут к их потомкам, родившимся в том порядке, как они расположены на рисунке, слева направо. Римскими цифрами (I, II и III) обозначены поколения, арабскими (I, 2 и 3) – члены семьи в каждом поколении. Для точного обозначения конкретного члена семьи используется двузначный код (например, II-3). Характерными признаками аутосомно-доминантного типа наследования являются: 1) симптомы заболевания проявляются в последующих поколениях в случае полной пенетрантности (т.е. если каждый генотип проявляется фенотипически); 2) лица мужского и женского пола поражаются с одинаковой частотой.
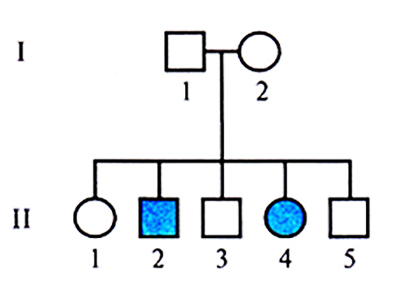
Рис. 20.2. Аутосомно-рецессивный тип наследования. Характерные признаки: 1) у здоровых родителей могут появляться больные дети; 2) лица мужского и женского пола поражаются с одинаковой частотой; 3) если больны оба родителя, то больны и все их дети (эта ситуация на рисунке на отражена).
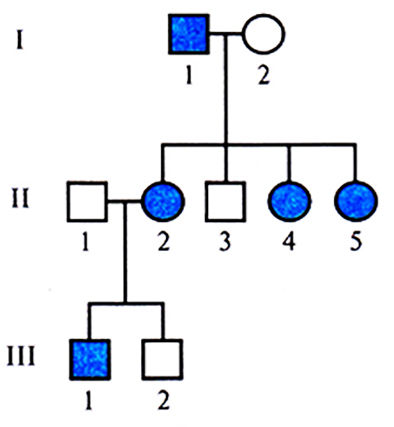
Рис. 20.3. Х-сцепленный доминантный тип наследования. Характерные признаки: 1) в случае полной пенетрантности больные присутствуют в каждом поколении; 2) больны все дочери пораженного мужчины, а все его сыновья здоровы; 3) в последующих поколениях часто проявляется тип наследования «от отца – к дочери – к ее сыну»; 4) число больных женского пола может быть больше, чем мужского.
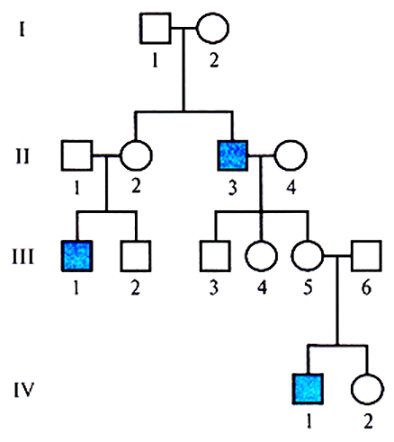
Рис. 20.4. Х-сцепленный рецессивный тип наследования. Характерные признаки: 1) у здоровых родителей могут рождаться больные дети; 2) нет прямой передачи заболевания от отца к сыну; 3) больных мужского пола больше, чем женского.
Клонирование генов человека не всегда состоит из строго определенных последовательных процедур. Скорее существует набор различных инструментов и способов, используемых в зависимости от конкретных условий. Например, начальный этап поиска гена, ответственного за данное заболевание, определяется наличием информации о его продукте. Как правило, при идентификации генов, ассоциированных с различными заболеваниями, нельзя обойтись без генетических и физических карт, а построение таких карт в конечном счете поможет определить нуклеотидную последовательность всего генома человека. Генетическая карта (карта сцепления) показывает расположение определенных сайтов (локусов) вдоль хромосомы. Для построения полных карт сцепления необходимо, чтобы локусы каждой хромосомы были представлены часто встречающимися аллелями и чтобы можно было легко идентифицировать каждый из них. Физическая карта – это набор упорядоченных клонов ДНК, охватывающих всю хромосому или какую-то ее область. На практике эти клоны перекрываются, образуя последовательность фрагментов, называемую контигом. Длина участка, охватываемого контигом, выражается в парах нуклеотидов. Физическая карта, состоящая из контигов, служит основой при построении окончательной физической карты, которая представляет собой полную нуклеотидную последовательность хромосомы.
ГЕНЕТИЧЕСКОЕ СЦЕПЛЕНИЕ И КАРТИРОВАНИЕ ГЕНОВ
В 1865 г. Грегор Мендель, основываясь на результатах своих опытов с садовым горохом, сформулировал основные принципы наследования признаков. Во-первых, он пришел к выводу, что единицы наследственности дискретны, встречаются парами и могут существовать в альтернативных формах. Позже (1905 г.) эти единицы назвали генами, а варианты одного гена – аллелями. Во-вторых, Мендель обнаружил, что в половую клетку (гамету) попадает только один ген из каждой пары. В-третьих, он заключил, что пары генов образуются независимо друг от друга, поэтому результатом единственного генетически значимого скрещивания будут все возможные генетические комбинации – в том случае, если число потомков достаточно велико (рис. 20.5). Последнее заключение, хотя Мендель и не знал этого, справедливо только для пар генов, находящихся на разных хромосомах или по крайней мере на разных концах одной хромосомы. В экспериментах Менделя ни при одном из скрещиваний не затрагивались такие пары генов, которые находились на одной хромосоме близко друг от друга. В противном случае он заметил бы, что эти гены наследуются не независимо, как сейчас говорят, они сцеплены.
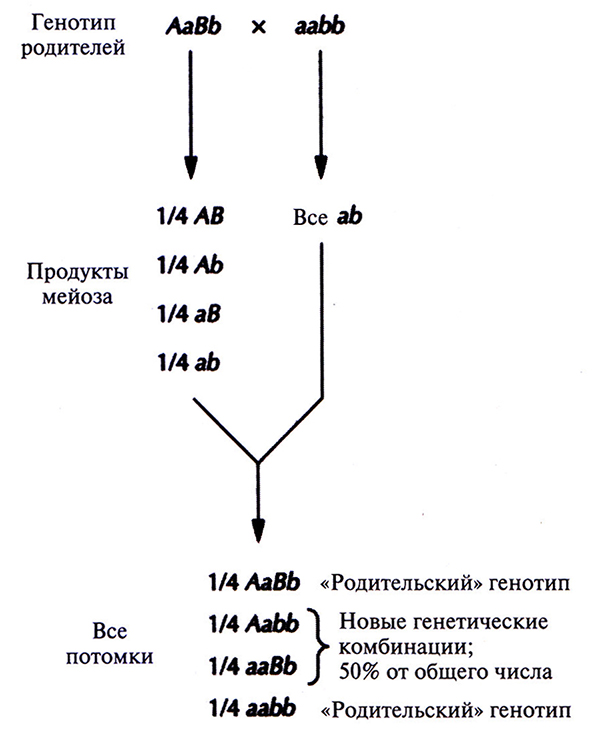
Рис. 20.5. Независимое распределение генов. При скрещивании дигетерозиготного индивида (АаВЬ) с индивидом, гомозиготным по двум рецессивным признакам (aabb), 50% потомков будут иметь родительские генотипы (АаВЬ, aabb), а 50% – новые комбинации генотипов (Aabb, ааВЬ) – в том случае, если число потомков достаточно велико для получения репрезентативной выборки. Этот результат означает, что гены А и В распределяются независимо, у потомков встречаются все возможные комбинации гамет каждого из родителей. Необходимо заметить, что процентное соотношение «родительских» и комбинированных типов зависит от генотипов родителей; например, в результате скрещивания особей с генотипами ААВВ и aabb потомки в 100% случаев будут иметь генотип, отличный от родительских (АаВЬ).
В принципе при полном генетическом сцеплении все гены любой хромосомы должны передаваться в половые клетки в виде неразделимых блоков, не образуя в процессе мейоза новых генетических комбинаций на хромосомах (рис. 20.6). Однако в большинстве случаев сцепление является неполным. При мейозе происходит обмен (рекомбинация, кроссовер) между генными сайтами (локусами), и создаются новые комбинации генов (рис. 20.7). Поскольку обычно рекомбинация происходит тем чаще, чем больше расстояние между двумя специфическими генными локусами, частоту рекомбинаций можно использовать как меру расстояния (генетического расстояния) между двумя генами. Таким образом, анализируя частоты рекомбинаций у потомков родителей, гетерозиготных по ряду сцепленных генов, можно построить генетическую карту, на которой гены будут расположены в линейном порядке. Расстояние между локусами отражает лишь частоту рекомбинаций и не эквивалентно точному физическому расстоянию. Однако, сравнивая физические и генетические карты хромосом, удалось установить соответствие между частотой рекомбинаций и числом нуклеотидиых пар ДНК. В качестве единицы при картировании используется 1 сантиморганида (сМ), величина, равная частоте рекомбинаций 1%, что для человека соответствует примерно 106 пар нуклеотидов (п. н.).
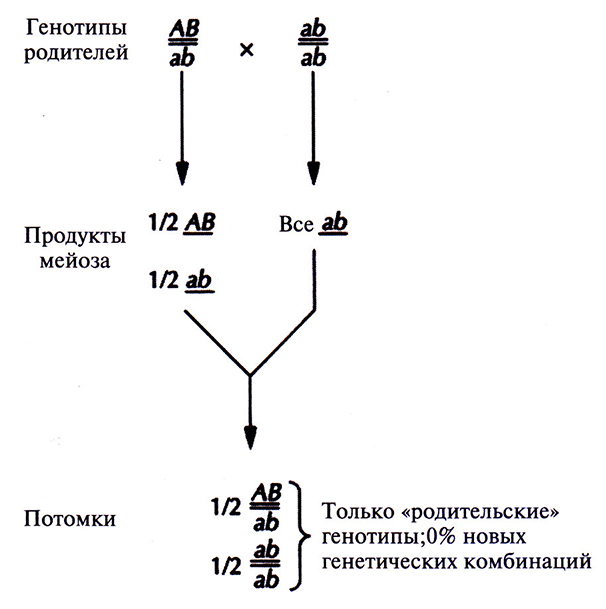
Рис. 20.6. Полное сцепление. Рассматриваемые аллели дигетерозиготного родителя, АВ//аЬ, находятся в фазе сцепления (цис), второй родитель гомозиготен по двум рецессивным признакам, его генотип ab//ab. В отсутствие рекомбинации между локусами А и В все потомки будут иметь родительские генотипы, половина – генотип АВ//аЬ и половина – аЬ//аЬ. Полное сцепление не всегда означает отсутствие новых генетических комбинаций; например, все потомки от скрещивания АЬ//аВ х АВ//аЬ будут иметь новые генетические комбинации, а именно: АЬ//АВ, АЬ//аЬ, аВ//АВ и аВ//аb. Однако в отсутствие рекомбинации гены одной и той же хромосомы будут всегда сцеплены вместе. Для удобства генетическая номенклатура использует одну горизонтальную или косую черту вместо двух для обозначения сцепления локусов пары одинаковых (гомологичных) хромосом.
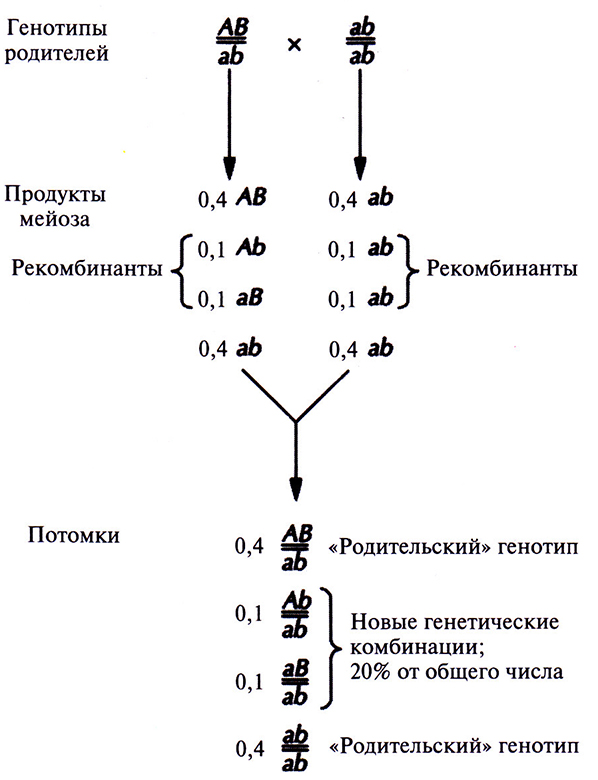
Рис. 20.7. Неполное сцепление. В данном примере 20% (т.е. 0,1 + 0,1 = 0,2) потомков имеют генотипы, сформировавшиеся в результате рекомбинации(й) между локусами А и В в процессе мейоза. Частота рекомбинаций не зависит от генотипов родителей. Родитель, гомозиготный по двум рецессивным признакам, производит только один тип гамет даже в случае рекомбинации. В анализирующем скрещивании рекомбинантные продукты мейоза проявляются у потомков фенотипически.
Отметим несколько важных моментов, касающихся генетического сцепления и картирования генов. Во-первых, чтобы можно было оценить частоту новых генетических комбинаций (рекомбинантов), один из родителей должен быть гетерозиготен как минимум по двум локусам (АВ/ab или Ab/аВ). Во-вторых, дигетерозиготные генотипы должны существовать в двух конфигурациях (фазах). Если два сцепленных гена на каждой из хромосом представлены одним типом аллелей (т.е. оба доминантные, АВ, или оба рецессивные, ab), то такую конфигурацию называют фазой сцепления (цис-фазой). Если же два сцепленных гена на каждой хромосоме представлены разными типами аллелей (т. е. один доминантный, а другой рецессивный, аВ или АЬ), то конфигурацию называют фазой отталкивания (транс-фазой). В-третьих, рекомбинация между двумя генами происходит независимо от их фазы. С точки зрения генетики рекомбинация между генами, находящимися в дигомозиготном состоянии (т.е. Ab/Ab или АВ/АВ), не приводит к появлению новой генетической комбинации, и поэтому, даже если подобная рекомбинация происходит, ее невозможно обнаружить. В-четвертых, частота рекомбинации 0% означает полное сцепление, а 50% – что гены расположены либо на разных хромосомах, либо на одной хромосоме, но удалены друг от друга слишком далеко для выявления сцепления. Для решения проблемы картирования двух сильно удаленных генов, расположенных на одной хромосоме, необходимо картировать гены, лежащие между ними, что позволит определить, образуют ли все они одну группу сцепления.
Для построения подробных генетических карт некоторых эукариотических организмов, таких как мышь, кукуруза, плодовая мушка, нематоды и дрожжи, необходимо идентифицировать целый ряд генов, каждый из которых представлен по крайней мере двумя аллелями. Затем нужно провести скрещивания и подсчитать частоту рекомбинаций у большого числа потомков. Результаты отражают степень сцепления между генами. В конце концов, используя мультифакторные (более двух пар сцепленных генов) скрещивания , можно получить детальные генетические карты.
ОБНАРУЖЕНИЕ И ОЦЕНКА
ГЕНЕТИЧЕСКОГО СЦЕПЛЕНИЯ У ЧЕЛОВЕКА
До появления в начале 1980-х гг. технологии рекомбинантных ДНК обнаружение и оценка генетического сцепления у человека представляли собой сложную и очень трудоемкую процедуру, которая к тому же обычно оказывалась безуспешной. При этом исследователи сталкивались с целым рядом проблем. Во-первых, генетический статус родителей обычно бывает неизвестен, что затрудняет разграничение рекомбинантных и нерекомбинантных потомков. Во-вторых, немногочисленность большинства семей снижает статистическую достоверность полученных результатов.
Наличие у мужчин одной Х-хромосомы значительно облегчает оценку генетических расстояний между генными локусами. В данном случае все аллели генов, расположенных на Х-хромосоме, проявляются фенотипически. Сыновья женщин, дигетерозиготных по Х-сцепленным локусам, получают рекомбинантную или нерекомбинантную Х-хромосому. Если фаза, в которой аллели двух генных локусов находятся у матерей, известна, то среди сыновей легко установить рекомбинантные и нерекомбинантные типы. Генотипы отцов в данном случае не имеют значения, поскольку сыновья наследуют только материнскую Х-хромосому. Иногда фазу аллелей у дигетерозиготной матери можно установить исходя из фенотипа ее отца. Например, если у отца матери (деда) два Х-сцепленных признака рецессивны, а у нее самой – доминантны, то мать дигетерозиготна, а рассматриваемые аллели находятся в цис-фазе, т.е. АВ/ab (рис. 20.8). Этот метод обнаружения сцепления основан на подсчете двухлокусных фенотипов у сыновей большого числа дигетерозиготных женщин с известной фазой аллелей. В этом случае доля хромосом, рекомбинантных по двум специфическим генным локусам (рекомбинационный индекс), будет равна сумме рекомбинантных хромосом (R), деленной на общее число хромосом – рекомбинантных и нерекомбинантных (NR):
S(R)/[S(R) + S(NR)].
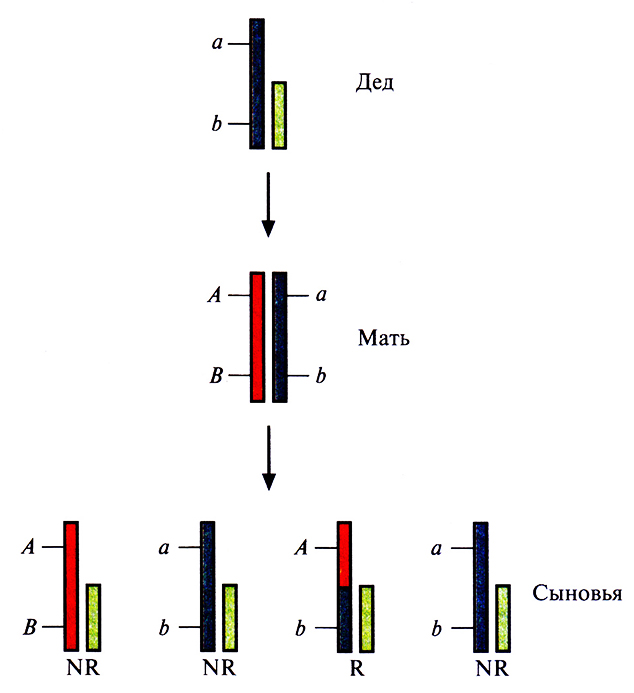
Рис. 20.8. Картирование Х-хромосомы. В этом случае генетическая фаза двух или большего числа Х-сцепленных локусов у дочери (Мать) устанавливается на основании данных о Х-сцепленных аллелях ее отца (Дед). Эту информацию в свою очередь используют для определения, какие из ее сыновей (Сыновья) получили рекомбинантную (R) и нерекомбинантную (NR) хромосому. В данном примере дед несет два рецессивных гена в локусах А и В Х-хромосомы, его дочь дигетерозиготна, а рассматриваемые аллели находятся у нее в цис-фазе. На Х-хромосоме показаны аллели локусов А и В, Х-хромосома изображена в виде более короткой полоски.
Однако данный подход имеет ряд недостатков. Во-первых, не всегда можно определить генотип деда, а следовательно, фаза, в которой находятся аллели у предположительно дигетерозиготной матери, остается неизвестной. Во-вторых, не все матери в большой выборке семей будут гетерозиготны по одним и тем же двум локусам. Несмотря на все усилия, до 1980-х гг. не удавалось построить достаточно протяженную однозначную карту сцепления Х-хромосомы человека, основанную на подсчете рекомбинантных и нерекомбинантных хромосом. В то время было известно всего несколько локусов и слишком мало аллелей было идентифицировано.
Анализ сцепления методом максимального правдоподобия:
логарифм соотношения шансов (лод-балл)
Кроме метода, в котором определяется частота рекомбинаций между двумя локусами на основании прямого подсчета рекомбинантных и нерекомбинантных хромосом, необходимо было разработать более общий, непрямой метод, который: 1) мог бы строго различать независимое распределение и сцепление; 2) не обязательно опирался бы на данные о фазе аллелей дигетерозиготных родителей; 3) мог суммировать информацию, полученную от большого количества различных семей; 4) позволял оценить рекомбинационный индекс в том случае, когда сцепление обнаружено. Такой метод, широко использующийся в настоящее время, был создан в 1955 г. Мортоном.
При изучении сцепления рекомбинационный индекс обозначается греческой буквой тета (⍬). В методе Мортона сравнивается вероятность L(⍬) того, что у братьев и сестер (сибсов) два локуса сцеплены (т. е. локализованы на одной хромосоме и находятся близко друг от друга), с вероятностью L(0,50) того, что два локуса не сцеплены (т.е. находятся на разных хромосомах или далеко друг от друга в пределах одной хромосомы), для любого рекомбинационного индекса ⍬. В случае сцепления, поскольку рекомбинационный индекс неизвестен, он может принимать любое значение в интервале от 0 до 0,5 (0 ≤ в < 0,50). Если же два локуса распределяются независимо, то ⍬ = 0,50 по определению. Другими словами, в том случае, когда половина гамет, полученных от дигетерозиготного родителя, содержит новые генетические комбинации, два локуса находятся либо на негомологичных хромосомах, либо настолько далеко друг от друга на одной хромосоме, что это выглядит так, будто они расположены на разных хромосомах. Следовательно, если L(⍬) = L(0,50), то два локуса не сцеплены. Десятичный логарифм отношения этих двух вероятностей, т.е. log[L(⍬ )/L(0,50)], представляет собой логарифм соотношения шансов (log-of-odds ratio), называемый лод-баллом (LOD). Лодд-балл обозначают буквой Z;Z(⍬) – это лод-балл для данного значения ⍬, где 0 ≤ ⍬ < 0,50.
L(⍬) можно определить, если известна вероятность получения конкретного сочетания рекомбинантных и нерекомбинантных хромосом для сибсов каждой изучаемой семьи. Вероятность того, что потомки получат от дигетерозиготного родителя нерекомбинантную хромосому, равна 1/2(1–⍬) + 1/2(1–⍬), или 1–⍬, а вероятность того, что они получат рекомбинантную хромосому, – 1/2⍬ + 1/2⍬, или ⍬. Например, в семье с пятью детьми вероятность для каждого из них получить нерекомбинантную хромосому от дигетерозиготного родителя составляет К(1–⍬)5, где (1–⍬) – вероятность получения нерекомбинантной хромосомы, показатель степени 5 – число сибсов с нерекомбинантной хромосомой, точнее, число нерекомбинантных хромосом у сибсов, К – коэффициент. Если все хромосомы одинаковы, т.е. все нерекомбинантные или все рекомбинантные, то К= 1 (т.е. 5!/5!0!, или n!/n!O!, где n – число сибсов в данной семье). В семье с четырьмя детьми вероятность того, что все они получат рекомбинантную хромосому от дигетерозиготного родителя, составляет ⍬4. Далее, вероятность того, что в семье с девятью детьми пять получат нерекомбинантные хромосомы и четыре – рекомбинантные, равна К(1–⍬)5(⍬)4, где К = 126, т.е. 9!/5!4!. Лод-балл выражается как отношение величин, имеющих одинаковые коэффициенты. Эти коэффициенты, стоящие в числителе и знаменателе, сокращаются, а потому при анализе сцепления не учитываются.
Проиллюстрируем подсчет лод-балла на примере сибсов одной семьи (рис. 20.9). Обозначения В и О на рис. 20.9 соответствуют аллелям АВО*В и АВ0*0 групп крови системы АВО. Закрашенными символами обозначено аутосомно-доминантное заболевание с полной пенетрантностью – наследственный онихоартроз (NPS, nail-patella syndrom). Основные признаки NPS – нарушение роста ногтей на пальцах рук и ног и редукция или отсутствие надколенника. Ген NPS обозначается NPS1, а его рецессивный («нормальный») и доминантный («патологический») аллели – NPS1*N и NPS1*D соответственно. NPS представляет собой подходящий для изучения сцепления признак, так как он рано диагностируется, не влияет на жизнеспособность и репродуктивную функцию и присутствует при рождении.
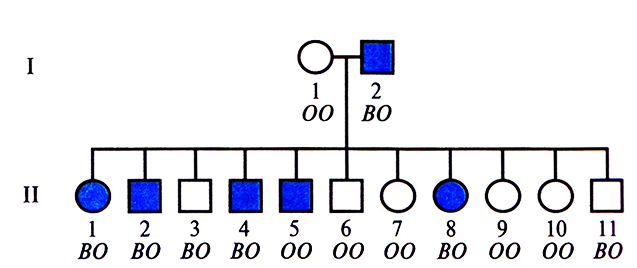
Рис. 20.9. Наследование генов онихоартроза и генов групп крови системы АВО. Закрашенными символами обозначены лица с наследственным онихоартрозом, незакрашенными – лица, у которых признаки данного заболевания отсутствуют Буквы под каждым символом обозначают аллели групп крови системы АВО (использованы сокращенные обозначения: О соответствует АВО*O, В – АВО*В).
Отец 1-2 (рис. 20.9) гетерозиготен по локусу NPS, поскольку среди его детей есть как больные, так и здоровые. Он гетерозиготен и по локусу АВО (АВО*В/АВ0*O), так как у его детей встречаются фенотипы О и В, а генотип его супруги (I-1) – АВO*O/АВ0*O. Следовательно, отец дигетерозиготен по этим двум аутосомным локусам (NPS1 *N/NPS1 *D; А ВО *В/АВО*O). Если локусы АВО и NPS1 сцеплены, то фаза, в которой находятся их аллели у отца, неизвестна (состояние с неизвестной фазой). Она может быть как АВО*В NPS1*D/AB0*O NPS1*N (фаза 1), так и АВО* В NPS1*N/ABO*O NPS1*D (фаза 2). Или, в сокращенном виде, В D/О N (фаза 1) или В N/O D (фаза 2).
Если предположить, что локусы АВО и NPS сцеплены и их аллели у отца находятся в фазе 1 (АВО*В NPS1*D/ABO*D NPS1*N), то дети II-1, II-2, II-4, II-6, II-7, 11-8, II-9 и 11-10 получили от него нерекомбинантную хромосому АВО*В NPS1*D или АВО*O NPS1*N (рис. 20.10). Все дети получили от матери (1-1) хромосому АВО*O NPS1*N, поскольку она гомозиготна по двум локусам (АВО*O NPS1*N/ABO*O NPS1*N). В данном случае генетический вклад матери известен и не влияет на анализ сцепления. Исходя из того, что рассматриваемые аллели у отца находятся в фазе 1, каждый из его детей II-3, II-5 и II-11 получил рекомбинантную хромосому. Следовательно, вероятность такого сочетания нерекомбинантных и рекомбинантных хромосом для данной семьи равна (1-⍬)8(⍬)3.
Рассматриваемые аллели у отца с такой же вероятностью могут находиться в фазе 2, т.е. А ВО*В NPS1*N/ABO*O NPS1*D. Тогда дети II-3, П-5 и II-11 получили от него нерекомбинантные хромосомы, а каждый из оставшихся детей унаследовал рекомбинантную хромосому (рис. 20.10). Вероятность такой комбинации для данной семьи равна (1–⍬)3(⍬)8.
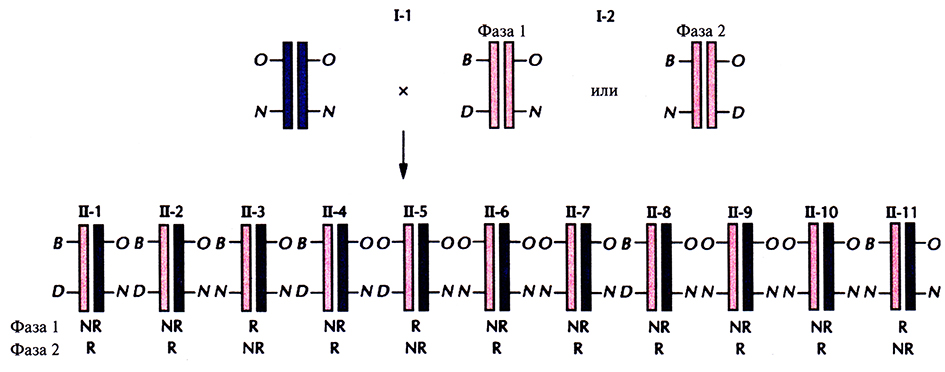
Рис. 20.10. Генетическая организация аллелей генов онихоартроза и групп крови АВО у членов родословной, приведенной на рис. 20.9, при условии сцепления этих двух локусов. Использованы сокращенные обозначения аллелей групп крови системы АВО: О соответствует АВO*O, а В – АВО*В. Рецессивный («нормальный») и доминантный («патологический») аллели локуса наследственного онихоартроза обозначены N и D соответственно. Генотип отца (1-2) может находиться в любой из двух фаз (фаза 1I, фаза 2). Хромосомы отца и хромосомы, унаследованные от него детьми, выделены синим цветом, хромосомы матери (I-1) и хромосомы, унаследованные от нее, – светло-коричневым Отмечено, какие из хромосом, полученных от отца, являются нерекомбинантными (NR) или рекомбинантными (R) для фазы 1 и фазы 2.
Поскольку для генотипа отца обе фазы равновероятны, общая вероятность L(⍬) наблюдаемой в родословной комбинации хромосом у его детей равна 1/2(1-⍬)8(⍬)3 + 1/2(1-⍬)3(⍬)8. Далее находят значение данного выражения для разных ⍬. Обычно используют следующий набор значений ⍬ : 0; 0,001; 0,05; 0,10; 0,2; 0,3; 0,4 и 0,50, а если нет ограничений во времени, можно взять весь спектр значений ⍬ от 0 до 0,50. Затем вычисляют логарифм отношения вероятности для каждого в, кроме 0,50, к вероятности для в = 0,50. Например, для ⍬ = 0,10 отношение L(0,10)/L(0,50) равно
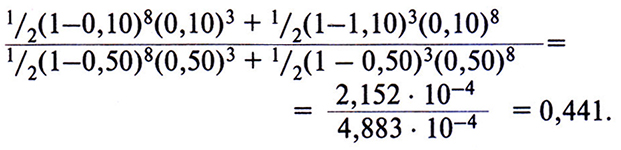
Десятичный логарифм 0,441 равен –0,356; это и есть лод-балл для данного отношения. Другими словами, Z(0,10) = –0,356.
Если фаза, в которой находятся рассматриваемые аллели у отца, известна, то и значение вероятности L(⍬) для данной семьи тоже будет известно. Например, если для генотипа 1-2 имеет место фаза 1 (АВО*В NPS1*D/ABO*O NPS1*N), то, как отмечалось выше, вероятность L(⍬) для данной семьи будет равна (1-⍬)8(⍬)3, и Z(⍬ = 0,10) составит
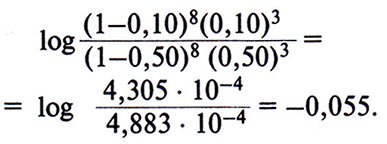
Если же для генотипа 1-2 имеет место фаза 2 (АВО*В NPS1*N/АВО*O NPS1*D), то Z(0,10) будет равен
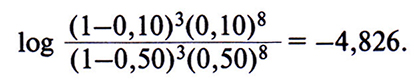
Для состояния с неизвестной фазой значения Z для родословной, приведенной на рис. 20.9, варьируют от –5,993 при ⍬ = 0,001 до +0,029 при ⍬ = 0,45 (табл. 20.1). Если сибсы получили хотя бы одну рекомбинантную хромосому и ⍬ = 0, то Z = – ∞. Как видно из табл. 20.1, лод-балл максимален (Zmax) при ⍬, близком к 0,30. Проведя дополнительные расчеты для ⍬ от 0,20 до 0,40, получим, что Zmax = +0,214 при ⍬ = 0,276.
Таблица 20.1. Значения Z при разных ⍬ для родословной, приведенной на рис. 20.9,
в случае состояния с неизвестной фазой

Значение Zmax = +0,214 не позволяет с уверенностью говорить о сцеплении локусов АВО и NPS1. Условились, что два аутосомных локуса могут считаться сцепленными только в том случае, если значение максимального лод-балла большее или равно +3,000: вероятность сцепления в этом случае составляет 1000 к 1 или выше. В случае Х-cцепленных генов, заведомо находящихся на одной хромосоме, значение Zmax, при котором можно говорить о сцеплении, больше или равно +2,000; это соответствует шансам в пользу сцепления 100 к 1 или выше. Если Z = –2,000, то сцепление двух локусов исключается, поскольку в этом случае в пользу сцепления существует лишь 1 шанс из 100.
Чтобы выявить сцепление, необходимо подсчитать Z-балл при разных ⍬ для разных семей и найти максимальное его значение. Преобразование отношения правдоподобий для каждой из семей в десятичный логарифм позволяет суммировать полученные Z(⍬). Для определения сцепления локусов АВО и NPS1 было проанализировано 25 родословных, в том числе несколько с большим количеством детей, и получено значение Z(0,10) = +31,235 (табл. 20.2); это больше, чем +3,000, следовательно, два указанных локуса сцеплены.
Значение ⍬, при котором Z достигает максимума, дает грубую оценку ре комбинационного индекса для двух сцепленных локусов. В первой работе по определению сцепления локусов АВО и NPS1 точное значение Zmax не определялось, но Z-балл при ⍬ = 0,10 был наибольшим из всех Z, подсчитанных для разных ⍬, из чего был сделан вывод, что, по-видимому, расстояние между этими двумя локусами составляет примерно 10 сМ. Необходимо подчеркнуть, что при анализе разных родословных с NPS обнаружилось, что с локусом NPS1 сцеплены разные аллели системы АВО. Другими словами, не существует специфического сцепления между конкретным аллелем системы АВО и локусом NPS1. Пока не доказано обратное, можно говорить только о генетическом сцеплении между локусами, а не между определенными аллелями. Следует также отметить, что метод подсчета лод-балла не позволяет определить аутосомную локализацию двух сцепленных локусов. Как мы увидим, для того чтобы установить, что локусы АВО и NPS1 расположены на длинном плече (q) хромосомы 9 между районами 34 и 34.2, т.е. 9q34–9q34.2, потребовались дополнительные исследования.
Таблица 20.2. Суммарные значения Z при разных ⍬ для локусов АВО и NPSI1)

ПОСТРОЕНИЕ ГЕНЕТИЧЕСКИХ КАРТ ХРОМОСОМ ЧЕЛОВЕКА
Генетический полиморфизм
Сцепление между локусом АВО и геном наследственного онихоартроза удалось обнаружить по двум причинам. Во-первых, каждый из основных аллелей системы АВО (IА, IB, IO) можно точно идентифицировать при помощи простого лабораторного теста, так что генотипы всех исследуемых родителей и детей оказываются известными. Во-вторых, каждый аллель системы АВО встречается в популяции с высокой частотой, и вероятность того, что родители будут гетерозиготны, достаточно высока. В Великобритании, где были проведены первые работы по изучению сцепления ABO-NPS, частоты аллелей IА, IB и IO составляют примерно 0,66; 0,28 и 0,06 соответственно.
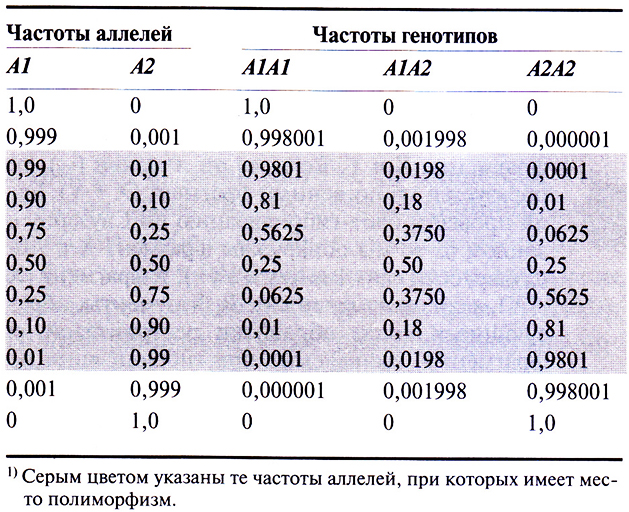
Термин «частота аллеля» обозначает долю конкретного аллеля среди всех аллелей данного локуса в популяции. Например, для двухаллельного локуса (А 1, А2) в популяции из 13 000 человек, где 3800 человек имеют генотип А1А1, 6400 – А1А2 и 2800 – А2А2, частота аллеля А1 составляет

Для большинства локусов частота одного аллеля (≥0,999) значительно превышает частоту другого (других) (≤0,001). Вследствие этого в больших популяциях подавляющее большинство (99,8%) особей оказываются гомозиготными по более часто встречающемуся аллелю, около 0,198% – гетерозиготными и 0,001% – гомозиготными по редкому аллелю. В подобных условиях практически невозможно установить сегрегацию аллелей данного локуса или его сцепление с другим локусом, поскольку большинство родителей будут гомозиготны по часто встречающемуся аллелю. Если же частоты двух аллелей данного локуса составляют 0,99 и 0,01, то гетерозиготными будут примерно 2% особей, и шансы обнаружить сегрегацию или сцепление возрастают, поскольку в популяции много особей, гетерозиготных по данному локусу (табл. 20.3). Таким образом, изучение сцепления у человека возможно только для локусов с часто встречающимися аллелями. Если два или больше аллелей данного локуса встречаются в популяции с частотой 0,01 и выше, то говорят, что имеет место генетический полиморфизм, и локус называют полиморфным. Поскольку генетический полиморфизм, подобный полиморфизму аллелей системы АВО, встречается редко, для осуществления проектов по картированию хромосом необходимо разрабатывать методы, которые позволяют с легкостью обнаруживать большое количество полиморфных сайтов.
Таблица 20.3. Частоты аллелей и генотипов
в большой популяции со случайным скрещиванием1)
Полиморфизм длины рестрикционных фрагментов
Для возникновения аллелей достаточно, чтобы два гомологичных гена различались всего одним нуклеотидом. Во многих случаях замена одного нуклеотида приводит к значительным различиям между продуктом измененного гена и нормальным белком. Однако множество однонуклеотидных замен не приводит к синтезу измененных генных продуктов, а кроме того, замены могут происходить в некодирующих областях ДНК и не приводить ни к каким последствиям. Такие «безвредные» замены, распределяясь по всей длине хромосомы, порождают полиморфные сайты (маркерные локусы, генетические маркеры), которые можно использовать для генетического картирования. Но сначала эти полиморфные сайты нужно обнаружить.
В 1980 г. Д. Ботштейн, Р. Уайт, М. Сколник и В. Дэвис (D. Botstein, R.L. White, М.Н. Skolnick, R.W. Davis) разработали теоретические основы идентификации однонуклеотидных полиморфных сайтов и использования их в качестве маркеров для построения хромосомных карт человека. Смысл методологии состоит в следующем. Рестрицирующие эндонуклеазы (рестриктазы) расщепляют ДНК в специфических сайтах. Когда однонуклеотидная замена происходит внутри такого сайта, рестриктаза перестает его расщеплять, но по-прежнему узнает и расщепляет интактный сайт в другой хромосоме (рис. 20.11, А). Поскольку один из аллелей содержит сайт узнавания для данной рестриктазы, а другой – нет, то при обработке ДНК этой рестриктазой образуются фрагменты разной длины. Наличие или отсутствие полиморфного сайта рестрикции можно установить, проведя гибридизацию ДНК с зондом, строго специфичным в отношении уникального участка хромосомы.
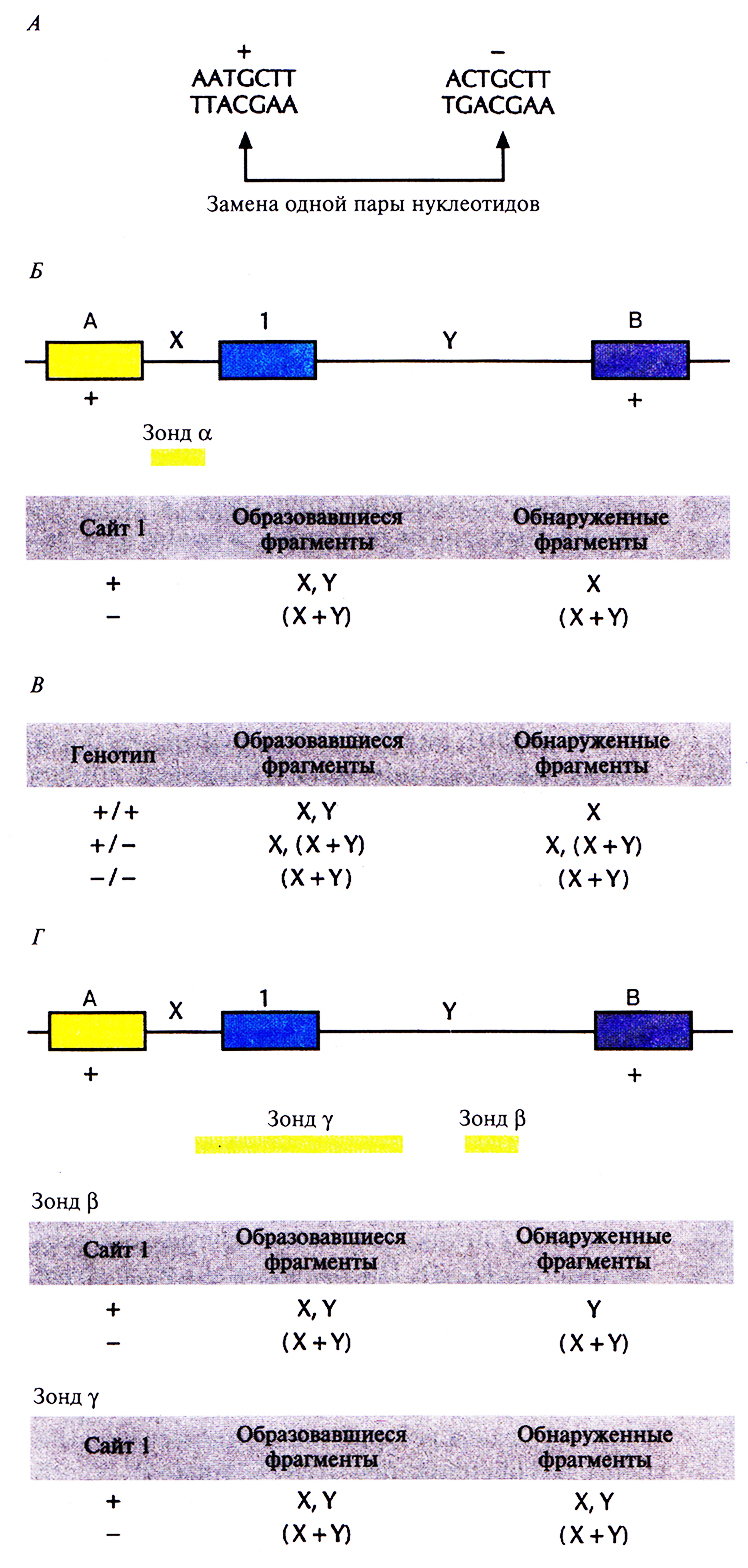
Рис. 20.11. Использование рестрикционных сайтов в качестве генетических маркеров. А. Замена одной пары нуклеотидов в рестрикционном сайте приводит к тому, что рестриктаза не распознает его и не расщепляет ДНК. Интактный и измененный сайты рестрикции отмечены знаками (+) и (–) соответственно. Б. Участок одной хромосомы, содержащий три сайта (А, 1 и В), узнаваемые одной и той же рестриктазой. X – расстояние между сайтами А и 1, Y – расстояние между сайтами 1 и В, X + Y – расстояние между сайтами А и В. Сайты А и В во всех случаях интактны (оба +), а сайт 1 может быть как интактным (+), так и измененным (–). Если сайт 1 интактен (+), то после обработки ДНК рестриктазой образуются фрагменты X и Y. Если же он изменен (–), то образуется единственный фрагмент (X + Y). Если провести блот-гибридизацию по Саузерну с зондом α, то мы обнаружим фрагмент X в том случае, если сайт 1 интактен (+), и фрагмент (X + Y), если он изменен (–). В. Фрагменты, образующиеся после обработки рестриктазой, и фрагменты, выявляемые при гибридизации по Саузерну с зондом α, для каждого из генотипов (+/+, +/–, –/–). Г. Фрагменты одной хромосомы, образующиеся после обработки рестриктазой, и фрагменты, выявляемые при гибридизации с зондом β или γ.
Предполжим, например, что какой-то участок хромосомы содержит три сайта, распознаваемых рестриктазой HindIll (рис. 20.11, Б), при этом у всех индивидуумов сайты А и В интактны. Это означает, что в популяции нет альтернативных аллелей по этим сайтам, т. е. отсутствует полиморфизм. В отличие от этого в сайте 1 с высокой частотой встречается однонуклеотидная замена, в результате чего он становится устойчивым к расщеплению HindIll .Таким образом, две хромосомы в популяции различаются по данному сайту: одна из них расщепляется (+), а другая нет (–).
Если расстояния от сайта А до сайта 1 и от сайта 1 до сайта В не совпадают, каждое из них не превышает 20 т. п. н. и существует однокопийный зонд, гибридизующийся с участком ДНК между сайтами А и 1 (рис. 20.11, Б), то после блот-гибридизации по Саузерну и разделения в агарозном геле фрагментов ДНК, полученных в результате обработки HindIll, мы сможем различить две ситуации. Первая – сайт 1 расщепляется, в результате чего образуется два фрагмента, и зонд гибридизуется с тем из них, который ограничивается сайтами А и 1. Вторая – сайт 1 не расщепляется и зонд гибридизуется с фрагментом ДНК, ограниченным сайтами А и В (рис. 20.11, Б).
Анализ реальных образцов ДНК несколько более сложен, поскольку хромосомы встречаются парами (рис. 20.11, В). Однако и в этом случае каждому генотипу (+/+,+/–, –/–) соответствует определенный набор фрагментов, образующийся в результате гибридизации с зондом. Кроме того, для выявления сайта рестрикции на участке 1 можно использовать зонды, гибридизующиеся с другими участками ДНК между сайтами А и В (рис. 20.11, Г). Феномен, состоящий в том, что наличие часто встречающегося в популяции измененного рестрикционного сайта приводит к образованию специфического набора фрагментов ДНК, называют полиморфизмом длины рестрикционных фрагментов (ПДРФ). Полиморфные сайты рестрикции образуют маркерные локусы на той хромосоме, где они присутствуют.
Генетический статус каждого ПДРФ-локуса на одной хромосоме называют гаплотипом. В случае одного сайта существуют два возможных гаплотипа (+ или –), в случае двух разных сайтов – четыре (++, + –, – + и – –); для n локусов число гаплотипов равно 2". Определение аллелей ПДРФ-локуса (или любых других полиморфных локусов), присутствующих на хромосомах данного индивидуума, называется гаплотипированием (генотипированием, ДНК-типированием). Наследование ПДРФ-локусов происходит в соответствии с законами Менделя, и можно проследить их передачу в пределах родословной. Если изучается наследование двух и более ПДРФ-локусов в данной семье, то можно выявить рекомбинацию. На рис. 20.12 представлена следующая ситуация: отец (I-1) гетерозиготен по трем разным ПДРФ-локусам, расположенным на одной хромосоме, а у матери (1-2) сайты рестрикции в трех рассматриваемых локусах отсутствуют. Генетический статус хромосомы, унаследованной от отца каждым из детей, можно установить путем генотипирования. Сын II-2 получил от отца хромосому, в которой произошел кроссинговер; остальные дети унаследовали от него нерекомбинантные хромосомы.
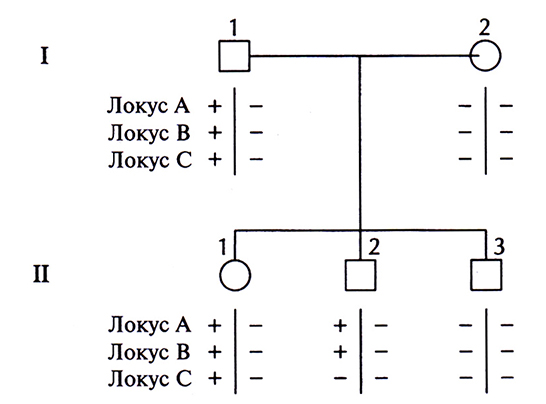
Рис. 20.12. Выявление сегрегации и рекомбинации ПДРФ-локусов в родословной. Знаками плюс (+) и минус (–) обозначены аллели, содержащие интактные и измененные сайты рестрикции, трех ПДРФ-локусов (А, В, С), расположенных на одной хромосоме. Генетический статус 1-1 определен исходя из генотипов его родителей, +++/+++ и – – –/– – – (не показаны). Определение гаплотипов у детей показывает, что II-2 получил от I-1 рекомбинантную хромосому, а II-1 и II-З – нерекомбинантные. Вертикальная черта, разделяющая наборы ПД РФ-аллелей под символом, обозначающим каждого члена семьи, разделяет гомологичные хромосомы.
На практике ДНК каждого индивидуума в отдельной пробирке обрабатывают различными рестриктазами, а затем гибридизуют с клонированными однокопийными фрагментами ДНК, которые используются в качестве зондов для выявления ПДРФ. Гибридизация ДНК-зонда с препаратом метафазных хромосом человека, распластанных на предметном стекле (гибридизация in situ), позволяет определить, соответствует ли данный зонд уникальному участку хромосомы (однокопийной ДНК). Для обозначения тысяч ПДРФ-локусов разработана стандартная номенклатура. Например, запись D21S18 соответствует локусу, идентифицированному с помощью ДНК-зонда (D), который гибридизуется с хромосомой 21 (21), представлен одной копией (S) и зарегистрирован Комитетом по систематизации карт сцепления человека (DNA Committee of the International System for Human Linkage Maps – ISLM) под восемнадцатым номером (18). Полиморфные маркерные сайты, расположенные внутри известных генов, обозначают по названию гена. Например, ADH – это полиморфный локус в гене алкогольдегидрогеназы (ADH1). Никакого стандартного обозначения для ПДРФ-аллелей не существует. В одних лабораториях аллели нумеруют по порядку (D1S34*1, D1S43*2 и т. д.), в других – по длине фрагмента (в т.п.н.), образующегося при наличии или в отсутствие сайта (сайтов) рестрикции (D4S56*8, D4S56*12, D4S56*4 ит. д.).
Уже идентифицированы тысячи ПДРФ-локусов, благодаря чему значительно увеличилось число аллелей, которые можно использовать для генетических исследований. Сцепление между ПДРФ-локусом (локуса ми) и геном того или иного заболевания можно установить, подсчитав «парный» («двухлокусный») лод-балл для гаплотипированных семей, в которых выявлены случаи изучаемого генетического заболевания. Аналогично, сцепление между ПДРФ-локусами можно обнаружить, проанализировав данные по родословным, представленным несколькими поколениями. Использование ПДРФ для картирования имеет несколько ограничений. Эти локусы распределены по хромосомам неравномерно, зонды в виде клонов поддерживать неудобно, а гаплотипирование большого числа индивидов из нескольких семей методом блот-гибридизации по Саузерну весьма трудоемко. К счастью, в геноме человека в большом количестве (>100 000) встречаются другие полиморфные локусы, содержащие простые повторяющиеся элементы из двух, трех или четырех пар нуклеотидов – короткие тандемные повторы (STR, от англ. short tandem repeats), которые легко регистрируются с помощью полимеразной цепной реакции (ПЦР).
Полиморфизм коротких тандемных повторов
По геному человека равномерно распределены примерно 100 000 блоков динуклеотидных повторов CA/GT [(СА) • (GT)] (рис. 20.13), содержащих от 1 до 40 повторяющихся CA/GT-элементов. Любой такой блок, локализованный в определенном участке хромосомы, передается из поколения в поколение с сохранением числа повторяющихся элементов. Для CA/GT-повтора принято обозначение (СА)n, где n – число СА-повторов. В геноме человека встречаются и другие динуклеотидные повторы [например, (АТ)n и т.д.], а также три-[(АТС)n и т. д.] и тетрануклеотидные [(ATCG)n и т. д.].
САСАСАСАСАСАСАСАСАСАСАСАСАСАСАСАСАСАСАСАСАСАСАСА
GTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGT
Рис. 20.13. Динуклеотидный тандемный повтор (СА)24, содержащий 24 повторяющихся элемента.
Чтобы идентифицировать полиморфные STR-локусы, нужно прежде всего провести скрининг геномной библиотеки человека, содержащей вставки небольшого размера (примерно 1000 п. н.), используя подходящий олигонуклеотидный зонд. Для идентификации клонированных (СА)n-повторов обычно используют зонд, состоящий из 15 СА-элементов. Каждую позитивную вставку секвенируют, чтобы установить длину СА-повтора и нуклеотидные последовательности фланкирующих его участков. Чтобы определить, являются ли фланкирующие последовательности однокопийными, проводят гибридизацию in situ с комплементарными им зондами, и если обнаруживается, что эти последовательности встречаются в геноме один раз, то синтезируют пару комплементарных им праймеров и проводят амплификацию СА-повтора. Далее, используя эту пару праймеров, проводят ПЦР-тестирование ДНК, полученной от большого числа индивидуумов. ПЦР-продукты, длина которых для удобства электрофоретического разделения выбирается примерно 200 п. н., разделяют в полиакриламидном геле. Если длина амплифицированного таким образом сегмента ДНК одинакова для всех образцов ДНК, значит, повтор не полиморфен (рис. 20.14, А), и наоборот, если образуются

Рис. 20.14. Типирование STR-локусов. А. ДНК, полученную от разных индивидов (n = 7), амплифицировали с помощью ПЦР, используя пару праймеров (X), фланкирующих (CA)•(GT)-noвтop. Размер всех образовавшихся ПЦР-продуктов одинаков (дорожки 1–7), следовательно, одинакова и длина STR. Судя по полученным данным, STR-локус представлен только одним аллелем. Б. То же, что и на рис. А, но с использованием другой пары праймеров (Y) для другого STR-локуса. Образование двух разных ПЦР-продуктов означает, что данный локус представлен двумя аллелями. Дорожки 1–3 соответствуют амплифииированному фрагменту ДНК индивидов, гомозиготных по одному STR-аллелю, дорожки 4 и 5 – амплифицированному фрагменту ДНК гетерозиготных индивидов, несущих два разных STR-аллеля, дорожки 6 и 7 – амплифииированному фрагменту ДНК индивидов, гомозиготных по другому STR-аллелю.
ПЦР-продукты разной длины, это указывает на полиморфизм по данному STR (STR-полиморфизм, STRP) (рис. 20.14, Б). Различающиеся по длине СА-повторы данного локуса представляют собой аллели (рис. 20.15). Такие аллели нередко встречаются с частотой 0,20 и даже больше.

Рис. 20.15. Два STR-аллеля. Один из них (аллель 1) содержит повтор (СА)15, другой (аллель 2) – (СА)10. Повторы в обоих случаях фланкированы одинаковыми уникальными последовательностями.
К настоящему времени уже обнаружены тысячи STRP-локусов. Для их обозначения используются те же правила, что и для ПДРФ-локусов. В то же время названия STRP-праймеров часто отличаются от названий локусов. Многие STRP-локусы были идентифицированы французскими исследователями при финансовой поддержке со стороны Фразцузской ассоциации по мышечным дистрофиям (Association Franchise contre les Myopathies), и это нашло свое отражение в том, что обозначения многих пар STRP-праймеров начинаются с аббревиатуры AFM, после которой идет идентификационный номер (AFM349xc5). Обозначение пары праймеров часто сопровождается обозначением соответствующего локуса [AFM349xc5 (D3S2017)].
В настоящее время для картирования генома человека используются в основном не ПДРФ-локусы, a STRP. В отличие от ПДРФ-зондов, которые необходимо клонировать в векторе, очищать и метить, в случае STRP-локусов нужна информация лишь о нуклеотидной последовательности пары праймеров, которая может храниться в компьютерной базе данных. Кроме того, STRP-локусы равномерно распределены в геноме человека; частоты STR-аллелей очень высоки, что обеспечивает высокую гетерозиготность, а сами аллели без труда идентифицируются после ПЦР-амплификации.
КАРТИРОВАНИЕ ЛОКУСА ГЕНЕТИЧЕСКОГО ЗАБОЛЕВАНИЯ
В ОПРЕДЕЛЕННОМ РАЙОНЕ ХРОМОСОМЫ
Анализ родословных не позволяет установить хромосомную локализацию гена того или иного заболевания, если только этот ген не находится на Х-хромосоме. Однако можно исследовать сцепление между геном данного заболевания и полиморфными ПДРФ- или STRP-локусами, идентифицируя последние с помощью соответствующих зондов. Этот подход дает наилучший результат в том случае, когда заболевание имеет четкие симптомы, его наследование носит однозначный характер и известна степень его пенетрантности.
Для анализа сцепления прежде всего берут пробы крови у членов нескольких семей, представленных двумя-тремя поколениями, либо у членов одной большой семьи, представленной несколькими поколениями, с данным генетическим заболеванием (при этом необходимо проинформировать всех испытуемых о целях анализа и получить их согласие). Клетки крови культивируют, что позволяет постоянно получать ДНК для дальнейших процедур без повторного забора крови. Проводят генотипирование ДНК каждого индивида по нескольким полиморфным маркерам. В некоторых исследованиях используют более 250 маркеров, представляющих разные участки всех аутосом. Для всех информативных семей для каждого полиморфного локуса и локуса генетического заболевания вычисляют двухточечный (двухлокусный) лод-балл. Если Z≥ +3,00, то имеет место сцепление, при Z≤ –2,00 сцепление исключается.
Для определения хромосомной локализации гена, ответственного за доброкачественные семейные судороги новорожденных (BFNC, benign familial neonatal convultions), была изучена большая семья, представленная несколькими поколениями, члены которой страдали данным заболеванием (рис. 20.16). Это состояние проявляется приступами неконтролируемых подергиваний мышц лица, туловища, рук и ног в первые шесть месяцев жизни. Примерно в 90% случаев симптомы исчезают после 1 года. По-видимому, припадки не оказывают влияния на неврологический и интеллектуальный статус. BFNC – редкое заболевание, которое имеет четкие клинические признаки, наследуется по аутосомно-доминантному типу и имеет высокую пенетрантность.
В исследованной родословной, включающей несколько поколений, два из всех протестированных полиморфных маркеров, D20S19 и D20S20, были сцеплены с локусом BFNC (табл. 20.4; рис. 20.16). Аллели локусов D20S19 и D20S20 каждого генотипированного члена родословной, представленной на рис. 20.16, обо значены числами, расположенными одно над другим. Верхние числа соответствуют аллелям локуса D20S19, нижние – D20S20. Вертикальная черта разделяет аллели локусов одной хромосомы.
Таблица 20.4. Двухлокусный лод-балл для локуса BFNC
и двух полиморфных локусов хромосомы 201)
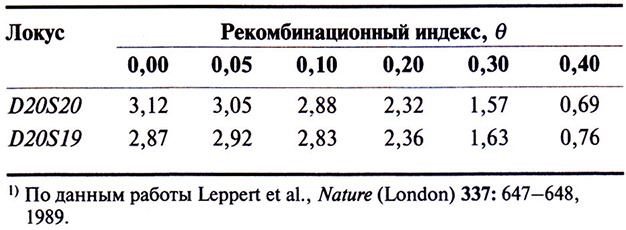
Рис. 20.16. Анализ гаплотипов двух хромосом по 20 полиморфным локусам в родословной, члены которой больны BFNC. Числа под символами – аллели двух полиморфных локусов, D20S19 и D20S20 (верхние и нижние числа соответственно). Гаплотипы двух хромосом индивидуума разделены вертикальной чертой. Перечеркнутые символы соответствуют умершим индивидам, цветные – липам, страдающим данным заболеванием. Символ с закрашенной четвертью квадрата отвечает случаю, когда «клинический» фенотип индивида отличался от фенотипов других больных членов семьи. II-8 и П-9 – неидентичные (разнояйцевые) близнецы. Символами со звездочкой отмечены случаи возможного проявления неполной пенетрантности (Ш-18, IV-4, IV-14). У больных членов семьи обнаруживается косегрегация гаплотипа (8,2) с заболеванием, все они несут эту хромосому, унаследованную от общего предка. (Поданным работы Leppert et al., Nature [London] 337: 647–648, 1989.)
С помощью зонда D20S19 было выявлено 10 аллелей ПДРФ-локуса, с помощью зонда D20S20 – 2 аллеля. В некоторых ПДРФ-локусах несколько сайтов узнавания для одной рестриктазы группируются на небольшом сегменте ДНК (примерно 20 т. п. н.) и все вместе рассматриваются как один локус. Четыре близкорасположенных сайта для одной рестриктазы могут приводить к образованию 20 фрагментов разной длины, которые выявляются одним зондом. STRP-локусы также могут иметь более двух аллелей.
Среди членов представленной на рис. 20.16 родословной в большинстве случаев наблюдается косегрегация гаплотипа (8,2) с заболеванием; это позволяет предположить, что в данной семье локус BFNC находится именно на хромосоме 8,2. Можно было ожидать, что и индивиды III-18, IV-4 и IV-14, получившие от больного родителя хромосому (8,2), также будут больны, однако это предположение не подтвердилось. Возможно, эти исключения связаны с неполной пенетрантностью заболевания. Другими словами, у индивидов III-18, IV-4 и IV-14 есть ген BFNC, но он не экспрессируется. Аналогичные случаи при анализе сцепления других заболеваний могут обусловливаться ошибками в диагностике или тем, что у некоторых индивидов, несущих ген заболевания, симптомы еще не проявились.
В нескольких случаях дети родителя, больного BFNC, несут хромосому (8,2) (например, IV-7 и IV-11), но никаких симптомов заболевания у них не обнаруживается. В обоих упомянутых выше случаях можно определить происхождение данной хромосомы. Например, индивид IV-7 унаследовал хромосому (14,1) от больного отца, а хромосому (8,2) с нормальным геном BFNC – от здоровой матери. Сложнее объяснить генотипы индивидов IV-9 и V-1. С одной стороны, они могли получить хромосому (8,2) с нормальным геном BFNC от здоровых предков.
ВАЖНАЯ ВЕХА
Построение генетической карты сцепления человека с помощью метода, основанного на полиморфизме длины рестрикционных фрагментов
D. Botstein, R. L. White, М. Skolnick, R. W. Davis Am. J. Hum. Genet. 32: 314-331, 1980
Часто встречающиеся типы полиморфизма у человека, которые можно типировать с помощью полимеразной цепной реакции
J. L. Weber, P. Е. May Am. J. Hum. Genet. 44: 388-396. 1989
Бурное развитие молекулярной генетики человека, начавшееся в 1980-х гг., стало возможным благодаря новаторским идеям Д. Ботштейна, Р. Уайта, М. Сколника и С. Дэвиса. Они обратили внимание, что полиморфизм длины рестрикционных фрагментов (ПДРФ) человека порождает полиморфные аллели (маркерные локусы), поддающиеся картированию. Как писали авторы в своей статье, «мы хотим предложить новый способ построения генетической карты сцепления человека. В его основе лежит создание при помощи технологии рекомбинантных ДНК случайных однокопийных ДНК-зондов, способных выявлять полиморфные нуклеотидные последовательности при гибридизации с индивидуальными ДНК, обработанными рестриктазой». Более того, они осознали, что, используя сцепление гена того или иного заболевания с маркерным локусом, можно определить хромосомную локализацию этого гена. Эта идея не была высказана ими прямо, но она непосредственно вытекала из их концепции. Ботштейн и др. пришли к абсолютно верному выводу: «Применение набора зондов, специфичных в отношении полиморфных участков ДНК, для анализа ДНК членов родословных с большим числом поколений откроет новые горизонты в генетике человека».
К 1992 г. на разных хромосомах человека были идентифицированы и картированы сотни ПДРФ-маркеров. С их помощью были изолированы гены таких наследственных заболеваний, как миодистрофия Дюшенна и муковисцидоз. К сожалению, высокополиморфные локусы расположены на разных хромосомах человека неравномерно и не всегда на близком расстоянии друг от друга. Кроме того, ПДРФ-анализ, основанный на гибридизации зонда с рестрицированной ДНК, весьма трудоемок и часто дает ошибочные результаты. Все эти проблемы удалось решить, когда Вебер и Мэй обнаружили, что по всему геному человека разбросано множество высокополиморфных ди-, три- и тетрануклеотидных повторов (коротких тандемных повторов; STS, от англ. short tandem repeats), вариации которых легко различаются при помощи ПЦР. Как писали авторы, «...данный тип полиморфных последовательностей, вероятно, найдет широкое применение при изучении многих генов наследственных заболеваний и позволит значительно увеличить разрешение генетических карт человека». STR, особенно динуклеотидные тандемные повторы, эффективны как маркеры; в этом качестве они уже вытеснили ПДРФ-локусы и в настоящее время используются для построения подробных генетических карт всех хромосом человека.
Например, индивид IV-9 мог унаследовать хромосому (8,2) через свою мать (III-15) от бабушки (II-6). С другой стороны, отсутствие у IV-9 и V-1 признаков заболевания может объясняться неполной пенетрантностью в том случае, если они унаследовали хромосому (8,2) с геном BFNC*D от больного родителя. Необходимо подчеркнуть, что в других семьях с BFNC может не наблюдаться сцепления аллелей D20S19*8 и D20S20*2 с аллелем данного заболевания. Так получилось, что в рассмотренном нами случае именно эти полиморфные аллели находятся на той же хромосоме, которая несет аллель BFNC*D и которая унаследована от одного предка. В обшем случае сцеплены локусы, а не аллели.
Из данных табл. 20.4 можно предположить, что расстояние от локусов D20S19 и D20S20 до локуса BFNC не превышает 5 сМ (<5 - 106п.н.). В общем случае анализ сцепления не позволяет разграничить два локуса, если расстояние между ними меньше 1–2 сМ. Поскольку локусы D20S19 и D20S20 расположены внутри района 13.2–13.3 длинного плеча (q) хромосомы 20 (20ql3.2-13.3), то и локус BFNC должен находиться вблизи данного района хромосомы или внутри него. К настоящему времени при помощи метода, основанного на вычислении лод-балла и использовании полиморфных маркеров, в специфических хромосомных участках было картировано более ста генов различных заболеваний.
ПОСТРОЕНИЕ МУЛЬТИЛОКУСНЫХ ХРОМОСОМНЫХ КАРТ ЧЕЛОВЕКА
Использование многих тысяч разбросанных по всему геному полиморфных маркеров позволило определять как порядок расположения локусов, так и расстояния между ними на каждой хромосоме. Карта сцепления полиморфных участков оказывается неоценимой при локализации генов различных заболеваний. Для идентификации таких генов можно использовать зонды, специфичные в отношении последовательностей, которые фланкируют данный ген.
Идеальными для картирования полиморфных локусов являются семьи, представленные тремя поколениями, в которых живы обе прабабки и оба прадеда, а родители имеют большое число детей (>8). Исходя из генотипов бабок и дедов, можно установить генетическую фазу, в которой находятся исследуемые локусы у каждого из родителей, а наличие большого числа детей повышает вероятность того, что рекомбинация произойдет. В Центре по изучению полиморфизма человека (СЕРН, Centre d'Etude du Polymorphisme Humain) в Париже собраны данные и образцы ДНК членов 65 семей, представленных в большинстве случаев тремя поколениями и имеющих в среднем по 8,5 детей (см., например, рис. 20.17). Этот банк семей (СЕРН-семей) предоставляет информацию о генотипах всех членов лабораториям всего мира, занимающимся картированием. В действительности он состоит из культур лимфобластозных клеточных линий большинства членов СЕРН-семей и служит готовым источником ДНК для картирования новых полиморфных локусов по мере их обнаружения.
Рис. 20.17. СЕРН-семья К1331.
Построение мультилокусной генетической карты (карты сцепления) хромосомы человека – непростая задача; для ее решения используют специализированную комьютерную программу, позволяющую установить порядок расположения локусов, наилучшим образом согласующийся с данными по рекомбинациям. Проблема упорядочивания локусов усложняется по мере возрастания числа локусов, которые необходимо картировать. Для N локусов сушествует N!/2 возможных вариантов их расположения. Так, для 10 локусов их число равно 1 814 400. И хотя некоторые комбинации заведомо нереальны, даже если основываться на визуальной проверке данных, все же число возможных вариантов остается очень большим. Обычно сначала находят наиболее вероятное расположение нескольких сцепленных локусов, а затем комбинируют эти «наилучшие» варианты и строят статистически достоверную карту сцепления всех локусов. Критерием того, расположен ли один локус рядом с другим, является значение десятичного логарифма правдоподобия (лод-балла); если он равен или превышает +3,00, то ответ будет положительным.
В общем случае построение карты проводят поэтапно. Сначала отбирают несколько полиморфных маркеров, расположенных на одной хромосоме. Потом генотипируют образцы ДНК, полученные от нескольких СЕРН-семей, по каждому полиморфному маркеру. Структура СЕРН-семей такова, что нет необходимости в определении генотипов всех образцов ДНК. Привлечение других семей не дает повышения качества карты, которое оправдывало бы дополнительную работу. Обычно используют 15, иногда – 40 семей. Для генотипирования 40 СЕРН-семей по 20 полиморфным маркерам необходимо провести примерно 10 000 анализов. Генотип каждого индивида по каждому локусу вводят в базу данных. На этом этапе происходит проверка базы данных на предмет ошибок. Компьютерная программа проводит поиск случаев несоответствия генотипов родителей и детей; эти ошибки возникают во время введения данных или генотипирования. Иногда для уточнения полученных результатов проводят повторное типирование. Ошибки могут приводить к неправильным выводам о расположении локусов и расстояниях между ними. Ошибочные данные по возможности исключаются из анализа. Для генотипированных СЕРН-семей определяют все «двухлокусные» лод-баллы и рекомбинационные индексы (⍬), и исходя из этих данных конкретная компьютерная программа строит генетическую карту (карту сцепления).
Карты сцепления хромосом человека постоянно обновляются по мере идентификации дополнительных полиморфных локусов. С увеличением числа локусов повышается разрешение карты и уменьшается расстояние между локусами. К 1994 г. были определены генотипы членов СЕРН-семей примерно по 6000 полиморфным маркерам и с помощью мультилокусного картирования установлено положение примерно 1000 локусов по всему геному человека со средним расстоянием между локусами около 4 сМ. Задача широкомасштабных проектов картирования состоит в том, чтобы, используя дополнительные полиморфные маркеры, построить карту каждой хромосомы с расстоянием между локусами 1–2 сМ.
Локализация гена заболевания на карте сцепления
Для решения этой задачи проводят генотипирование членов семей с определенным генетическим заболеванием по полиморфным маркерам, которые, по данным картирования, находятся на том же плече хромосомы, что и ген заболевания. Используют те же подходы, что и при вычислении двухточечного лод-балла при анализе сцепления. В данном случае локус гена заболевания произвольно размещают среди четырех упорядоченных локусов и вычисляют лод-балл для каждой позиции. В случае мультилокусного картирования лод-балл равен логарифму отношения 1) вероятности того, что ген заболевания занимает определенное положение на карте из четырех упорядоченных локусов, к 2) вероят но сти того, что ген заболевания не сцеплен ни с одним из рассматриваемых полиморфных локусов. Использование именно четырех полиморфных локусов обусловлено тем, что при большем их числе слишком сильно усложняются расчеты. Ген заболевания может располагаться до первого локуса, в разных областях между локусами или за последним локусом. Рассчитав лод-балл для каждого положения гена, которое он может занимать в различных наборах из четырех локусов, выбирают максимальное его значение, превышающее +3,00; оно дает наиболее вероятную локализацию данного гена.
Картирование с использованием радиационных гибридов
Для картирования с использованием радиационных гибридов (РГ-картирования) не нужно собирать родословные и генотипировать членов банка СЕРН-семей. В основе метода лежит работа с соматическими клетками и скрининг (с использованием ПЦР-зондов) клеточных линий, содержащих части (фрагменты) хромосом человека. РГ-картирование целой хромосомы или какой-то ее области начинается с создания гибридной (человек/грызун) клеточной линии, содержащей одну хромосому человека. Клетки такой монохромосомной гибридной клеточной линии подвергают воздействию летальных доз ионизирующей радиации (рентгеновских или гамма-лучей), в результате чего разрушаются клеточные мембраны, инактивируются ферменты, происходит фрагментация хромосом. Единицей измерения дозы ионизирующего излучения, поглощенной биологическим объектом, является рад (rad, от англ. radiation absorbed dose). Один рад равен 0,01 Дж на 1 кг ткани или 100 эргам на 1 г ткани. Обычно клетки в культуре погибают при 3000 рад. Чем больше доза, тем более сильные повреждения возникают и тем меньше размер образующихся фрагментов ДНК. При дозе 10 ООО рад фрагменты слишком малы для РГ-картирования.
Очень важным моментом при РГ-картировании является высвобождение и сохранение фрагментов ДНК человека, полученных после облучения. Чтобы решить эту задачу, проводят слияние облученных (донорских) клеток с необлученными (реципиентными) клетками грузынов. Облученные клетки, слившиеся друг с другом или оставшиеся изолированными, не способны расти в культуре вследствие радиационных повреждений. В свою очередь, реципиентные клетки, как слившиеся друг с другом, так и не слившиеся, лишены селективного маркера, который присутствует в донорских клетках и обеспечивает их рост в культуральной среде, используемой для слияния. Следовательно, в данной среде будут пролиферировать лишь слившиеся клетки донор–реципиент, несущие селективный маркер, при этом большинство фрагментов ДНК облученных клеток окажутся встроенными или транслоцированными на функциональные хромосомы реципиентных клеток. Выжившие слившиеся клетки культивируют вместе до тех пор, пока не установятся отдельные клеточные линии – так называемые радиационные гибриды (РГ). Группу радиационных гибридов, полученных в результате одного эксперимента, называют панелью радиационных гибридов (РГ-панелью). В ней в виде фрагментов хранится большая часть хромосомной ДНК человека, полученной из монохромосомной клеточной гибридной линии.
ДНК каждого члена РГ-панели анализируют с помощью нескольких хромосомоспецифичных ПЦР-зондов, многие из которых «узнают» полиморфные участки. Однако полиморфизм как таковой не требуется для РГ-картирования. Цель такого картирования – выяснить, присутствует ли данный участок хромосомы в клеточных линиях РГ-панели. Следовательно, для скрининга можно использовать и ПЦР-праймеры, специфичные в отношении уникальных (однокопийных) последовательностей ДНК. Мономпрфные ПЦР-идентифицируемые хромосомоспецифичные участки называют ДНК-маркирующими сайтами (STS, от англ. sequence /agged sites). Все клеточные линии РГ-панели проверяют на наличие (+) или отсутствие (–) такого сайта (табл. 20.5), используя весь набор зондов, и гибриды, ДНК которых не амплифицируется, отбраковывают. Для эффективного РГ-картирования необходима панель примерно из 100 РГ, полученных из одной монохромосомной гибридной клеточной линии.
Таблица 20.5. Данные по сохранению маркеров при РГ-картировании1)

Теоретическая основа РГ- и мейотического картирования весьма сходна. Чем ближе друг к другу на хромосоме находятся два участка, тем выше вероятность того, что оба они окажутся в одном фрагменте ДНК после облучения. Точно так же, чем ближе друг к другу находятся сайты, тем с большей вероятностью они не разойдутся при мейотическом картировании в результате рекомбинации. Основные положения, на которых базируется РГ-картирование, состоят в следующем: 1) индуцированный облучением разрыв между двумя сайтами не зависит от сохранения маркера; 2) сохранение фрагмента с одним маркером не зависит от сохранения любого другого фрагмента в этой же клетке.
Полученные для всех клеточных линий РГ-панели паттерны (паттерны сохранения, сигнатура) наличия (+) или отсутствия (–) каждого маркера используют для построения РГ-карты. Лод-балл рассчитывают как логарифм отношения вероятности получения конкретного паттерна сохранения двух сайтов к вероятности того, что при облучении эти сайты всегда разделяются разрывом. В отличие от мейотической рекомбинации, ⍬ для частоты радиационных разрывов принимает значения от 0 до 1; ⍬ = 0 означает, что два маркерных сайта никогда не разделяются при определенной дозе облучения, т.е. они тесно сцеплены При ⍬ = 1 маркеры всегда разделяются при определенной дозе облучения, т. е. вообще не сцеплены. Если лод-балл равен или больше +3,00, можно с уверенностью говорить о сцеплении двух маркеров. Разработаны компьютерные программы, позволяющие упорядочивать сайты и определять расстояние между ними на РГ-карте.
Расстояние между сайтами на РГ-карте измеряется в так называемых сантирэях (сР). Поскольку размер фрагментов обратно пропорционален дозе облучения, необходимо указывать дозу, при которой была получена данная РГ-панель и построена РГ-карта. Например, расстояние в 1 сР8000 означает, что при дозе 8000 рад между двумя маркерами происходит разрыв в 1 % случаев.
Прямой связи между сантирэями и числом пар нуклеотидов не существует. Можно лишь сказать, что чем выше доза в радах, тем меньше физическое расстояние для конкретной величины в сантирэях. Например, расстояния в 1 сP9000, 1 сР8000, сР6000, 1 сР5000, и 1 сРЗ000 эквивалентны примерно 50, 53, 62, 90 и 100 т.п.н. соответственно. Мейотическое же (генетическое) картирование способно дифференцировать сайты, находящиеся на расстоянии друг от друга в лучшем случае 1 сМ, т.е. 1000 т. п. н. РГ-карты не только имеют более высокое разрешение, но и являются более полными, чем генетические. Кроме того, РГ-картирование проще мейотического в техническом плане, а новые сайты можно быстро включать в ранее построенную РГ-карту. К сожалению, РГ-картирование не позволяет локализовать гены тех или иных заболеваний в специфических районах хромосомы. Несмотря на это при построении мультилокусных карт хромосом человека РГ-картирование, вероятно, вытеснит картирование по сцеплению, основанное на генотипировании членов СЕРН-семей.
ФИЗИЧЕСКОЕ КАРТИРОВАНИЕ ГЕНОМА ЧЕЛОВЕКА
Генетические и РГ-карты указывают линейное расположение маркерных сайтов. Расстояния между сайтами измеряются в условных единицах, которые отражают частоту рекомбинации (сМ) или вероятность сохранения двух сайтов в одном радиационном гибриде (сР). Эти единицы можно перевести (в некотором приближении) в единицы реальных физических расстояний – пары нуклеотидов, которые используются в физических картах. Физическая карта целой хромосомы или ее области дает непосредственное представление о расположении генов в ДНК, что облегчает их идентификацию и характеристику и систематическое секвенирование хромосомной ДНК.
Для построения физической карты необходимо прежде всего выделить из библиотеки геномной ДНК клоны, содержащие перекрывающиеся сегменты. Исходя из данных о перекрывающихся участках и другой информации о положении клонов, можно реконструировать непрерывный ряд клонированных сегментов какого-то района хромосомы, целой хромосомы или всего генома. Были получены упорядоченные наборы смежных (contiguous) клонов (контиги) на основе YAC-(yeast artifitial chromosome, искусственная хромосома дрожжей), ВАС-(bacterial artifitial chromosome, искусственная хромосома бактерий), PAC-(bacteriophage PI artifitial chiomosome, искусственная хромосома бактериофага PI) и космидных библиотек ДНК человека. Отметим, что стратегия построения контигов из крупных фрагментов ДНК человека, содержащихся в YAC-, ВАС- или РАС-библиотеках, немного отличается от таковой для Р1- или космидных контигов.
Построение контигов из YAC-, ВАС- и РАС-библиотек
При построении физических карт тех или иных районов хромосом или целых хромосом из геномных библиотек, содержащих крупные вставки (YAC-, ВАС- или РАС-библиотек), наиболее приемлем метод картирования, основанный на использовании STS. STS – это короткий однокопийный участок ДНК (примерно 100–300 п. н.), который можно выявить при помощи ПЦР с использованием уникального набора праймеров. Для получения протяженного контига, охватывающего значительный участок хромосомы, требуется большое число STS, находящихся на расстоянии 50–100 т.п.н. друг от друга. Например, для физического картирования хромосомы длиной примерно 200 миллионов пар нуклеотидов (м.п.н.) необходимо от 1500 до 3000 STS. Для построения же достаточно точной физической карты всего генома человека их нужно по меньшей мере 30 000.
Для создания STS были разработаны различные подходы. В одном из них ДНК из очищенного препарата одной хромосомы человека, изолированной при помощи проточной цитофотометрии, обрабатывают рестриктазой и клонируют в векторе, способном акцептировать небольшие (< 1000 п.н.) фрагменты ДНК. Затем секвенируют вставки из клонов, выбранных случайным образом, и отбрасывают те клоны, в которых вставки короче 100 п.н., и те, которые содержат последовательности из повторяющихся элементов ДНК человека. Наличие повторов определяют при помощи компьютерных программ, сравнивая нуклеотидную последовательность вставки с последовательностями всех известных повторов ДНК человека. Затем для каждого отобранного клона находят нуклеотидные последовательности праймеров. Каждый STS тестируют на предмет уникальности амплифицируемого фрагмента хромосомной ДНК.
С помощью ПЦР-скрининга выявляют STS в индивидуальных клонах библиотеки с крупными вставками, а затем, основываясь на распределении STS в клонах, численными методами находят вероятный набор перекрывающихся клонов и относительное положение имеющихся STS (рис. 20.18). С разработкой метода РГ-картирования появилась возможность без особого труда упорядочить STS, что облегчает идентификацию составляющих контига (рис. 20.19). Выявив перекрывающиеся клоны, определяют степень их перекрывания, размер контига и общую длину охватываемой им ДНК, с помощью эндонуклеазного картирования с использованием электрофоретической системы, разделяющей фрагменты ДНК длиннее 105 п.н. (например, импульсный электрофорез). Уже получены контиги хромосомных районов, охватывающие от 1 до более чем 20 м.п.н., а в ряде случаев – и целые хромосомы. В конце концов будут получены контиги из крупных фрагментов ДНК, перекрывающие весь геном.
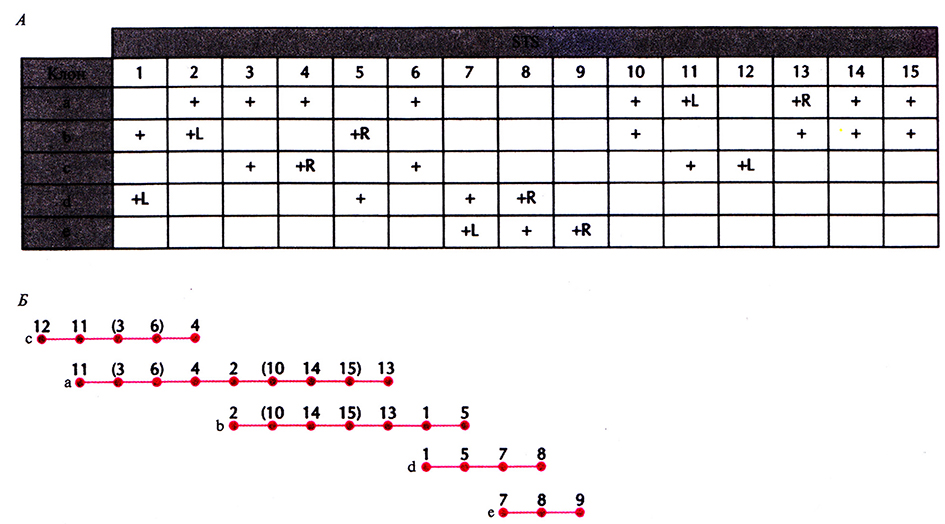
Рис. 20.18. STS-картирование. A. STS (с 1 по 15), обнаруженные с помощью ПЦР-скрининга в клонах а–е, обозначены знаком плюс (+). Буквами L и R указаны STS, находящихся на 5'- и 3'-концах вставки (на ее левом и правом концах соответственно). Б. Идентифицировав перекрывающиеся участки, можно построить контиг из пяти клонов и карту расположения STS. Полученные данные не позволяют установить порядок некоторых из них (номера в скобках). Интервалы между STS представлены одинаковыми; в действительности они неизвестны.
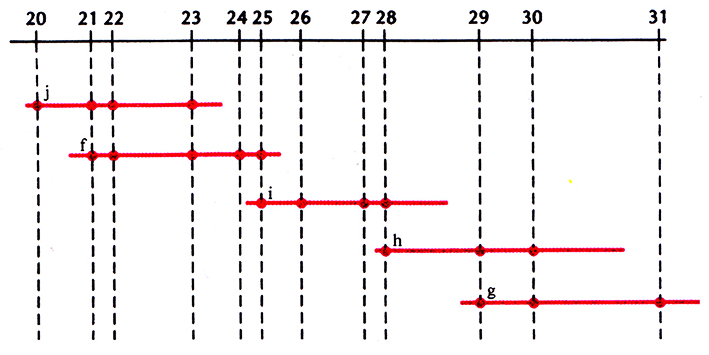
Рис. 20.19. Картирование с помощью упорядоченных STS (числа с 20 по 31 над горизонтальной линией). Точками указан STS-состав клонов f–j. STS упорядочены, что позволяет без труда идентифицировать перекрывающиеся клоны.
Построение контигов из космидных, Р1- и λ-библиотек
Более удобными для генетических исследований и широкомасштабного секвенирования часто оказываются контиги из небольших фрагментов ДНК, чем из крупных. Для построения контигов определенных районов хромосом или целых хромосом нередко используют космидные библиотеки. Обычно перекрывающиеся космидные клоны идентифицируют методом геномной дактилоскопии. Для этого из каждого клона экстрагируют ДН К и обрабатывают ее рестриктазой. Полученные фрагменты метят, разделяют при помощи электрофореза и визуализируют радиоавтографическими методами. Каждый клон порождает специфический набор фрагментов – уникальный отпечаток его ДНК; у перекрывающихся клонов один или несколько фрагментов совпадают.
Для концевого мечения ДНК-фрагментов – независимо от характера образующихся после эндонуклеазной обработки концов (5'-, З'-выступающих или тупых) – можно использовать реакцию замещения, катализируемого ДНК-полимеразой Т4. В этом случае к препарату космидной ДНК, обработанной рестриктазой, добавляют ДНК-полимеразу и один меченый дезоксинуклеотид (рис. 20.20). Под действием З'-экзонуклеазной активности ДНК-полимеразы происходит последовательное отщепление 3'-концевых нуклеотидов. Процесс продолжается до тех пор, пока в противопложной цепи не экспонируется нуклеотид, комплементарный меченому дезоксинуклеотиду, добавленному в реакционную смесь. Далее включается полимеразная активность ДНК-полимеразы, и к 3'-концу присоединяется свободный меченый нуклеотид. Поскольку другие нуклеотиды в реакционной смеси отсутствуют, дальнейшего роста цепи не происходит.
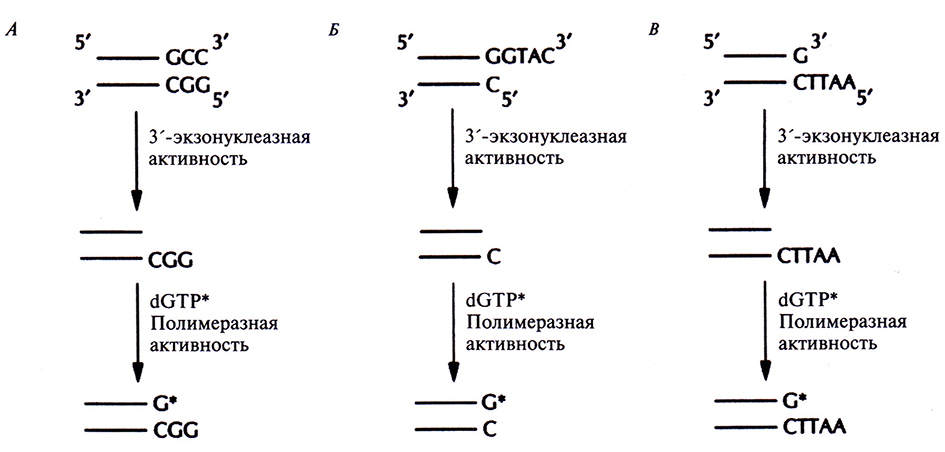
Рис. 20.20. Концевое мечение двухцепочечных ДНК с помощью ДНК-полимеразы Т4. 3'-экзонуклеазная активность ДНК-полимеразы катализирует отщепление З'-концевых нуклеотидов фрагментов ДНК с тупыми концами (А), с выступающими З'-концами (Б) или с выступающими 5'-концами (В). Отщепление происходит до тех пор, пока на противоположной цепи не экспонируется основание, комплементарное меченому дезоксирибонуклеотиду, введенному в реакционную смесь (dGTP*); затем «включается» полимеразная активность ДНК-полимеразы Е4, и к З'-концу присоединяется свободный меченый дезоксирибонуклеотид. Метка включается в оба конца фрагментов ДНК (на рисунке это не показано). Для мечения можно использовать любой дезоксирибонуклеотид.
Для мечения эндонуклеазных фрагментов существуют и другие способы. Усеченный З'-конец фрагмента можно удлинить (достроить) при помощи фрагмента Кленова, использующего выступающий 5'-конец в качестве матрицы; достраивание осуществляется за счет добавленных в реакционную смесь дезоксирибонуклеотидов, один из которых несет метку. Кроме того, к выступающим концам рестрикционных фрагментов можно присоединять меченые линкеры.
Для выявления перекрывающихся участков необходимо проанализировать очень большое число космидных клонов, поэтому для поиска меченых рестрикционных фрагментов, общих для пары клонов, используют специальные компьютерные программы. ДНК-отпечаток каждого клона сканируют, информацию вводят в компьютер и проводят попарные сравнения. По результатам этих сравнений организуют из клонов контиги. Наличие перекрывающихся участков в клонах созданного контига подтверждается построением подробных рестрикционных карт вставок. Пробелы между контигами заполняют, выбирая зонды из ближайших концов соседних контигов и проводя скрининг библиотеки, содержащей крупные вставки, для поиска недостающих участков ДНК.
Транскрипционное картирование
Клоны кДНК-библиотеки представляют собой ДНК-копии тех транскриптов экспрессирующихся генов, которые присутствовали в конкретной ткани в момент экстракции из нее мРНК. «Привязка» индивидуальных клонов кДНК к хромосомным районам создает предпосылки к выявлению возможных кандидатов на роль генов тех или иных заболеваний. Если анализ генетического сцепления показывает, что ген данного заболевания находится в той же области хромосомы, что и кДНК-последовательность(ти), то можно проверить, не происходят ли данные клоны кДНК из гена этого заболевания.
Привязку кДНК-клонов и других типов нуклеотидных экспрессируемых последовательностей к специфическим хромосомным районам называют транскрипционным картированием Для построения транскрипционных карт используют различные методы. В одном из них частично секвенируют отдельные кДНК-клоны и на основе транслированной части каждой кДНК-вставки получают STS. Данный тип STS называют экспрессируемым STS (eSTS). Для определения хромосомной локализации eSTS используют линии соматических гибридных клеток, которые содержат единственную хромосому человека (монохромосомные гибриды) или фрагменты конкретной хромосомы человека (делеционную панель). Для этого ДНК каждого из монохромосомных гибридов амплифицируют методом ПЦР с использованием eSTS-праймеров и идентифицируют ту хромосому, которая содержит данный eSTS. Затем методом ПЦР-амплификации ДНК клеточных линий делеционной панели идентифицируют район хромосомы, в котором находится данный eSTS (рис. 20.21). Кроме того, внутригенные STS можно нанести на существующие РГ карты. К 1996 г. на всех аутосомах и Х-хромосоме было картировано около 20 000 внутригенных STS человека.
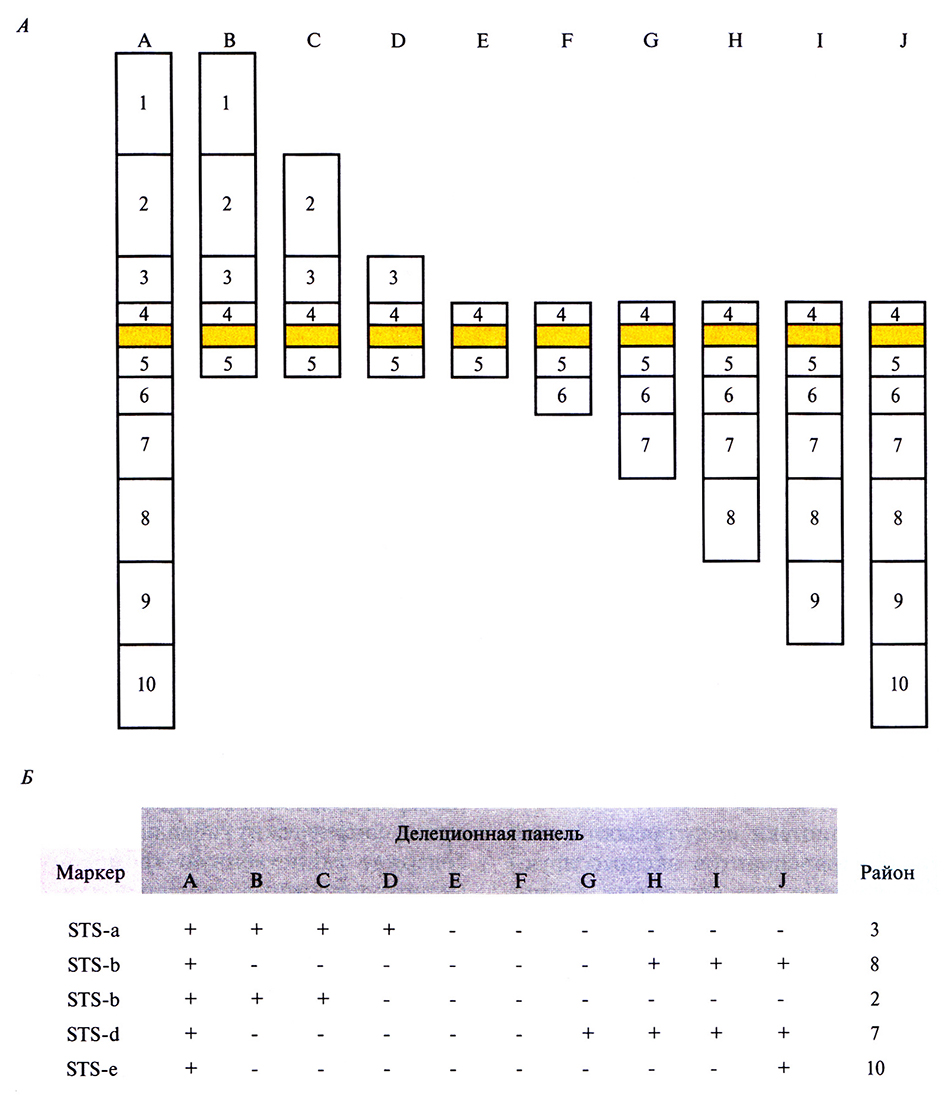
Рис. 20.21. Привязка маркера к специфическому хромосомному району с использованием делеционной панели гибридных клеток. А. Схематическое представление районов хромосомы, присутствующих в монохромосомном клеточном гибриде (А) и в клеточных линиях В–J делеционной панели гибридных клеток. Районы (с 1 по 10) определяются границами делеций в хромосомах делеционной панели. Закрашенный прямоугольник – центромера каждой хромосомы. Б. Результаты ПЦР-амплификации ДНК гибридных клеточных линий (A–J) с использованием STS-маркеров (с STS-a по STS-e). Наличие или отсутствие ПЦР-продукта указано знаком плюс или минус соответственно. Данные о наличии или отсутствии ПЦР-продуктов клеточных линий делеционной панели с STS-маркером используются для определения хромосомного района, в котором находится STS. Например, STS-d отнесен к району 7, поскольку соответствующий ПЦР-продукт образуется при амплификации каждой клеточной линии делеционной панели, в которой присутствует район 7.
Помимо экспериментов по картированию кДНК-клонов в хромосомных районах, в Институте исследования генома в Роквилле, Мэриленд (The Institute for Genome Research) и других лабораториях приступили к реализации проекта по частичному секвенированию клонов кДНК-библиотек всех органов и тканей человека. Одной из задач этой программы является создание каталога коротких последовательностей (150–300 нуклеотидов) для каждого экспрессируемого гена человека. Такие короткие кодирующие последовательности называют маркерными экспрессируемыми последовательностями (EST, от англ. expressed sequence tags). С их помощью можно изучать размеры, разнообразие и транскрипционную активность экспрессирующихся генов человека. Более того, на основе EST можно создавать STS и использовать их для картирования и отбора геномных клонов, содержащих данный ген.
Частичное секвенирование кДНК-клонов и обработка полученных данных полностью автоматизированы. Каждую новую EST сравнивают с теми, которые уже были секвенированы, и если последовательность действительно является новой, ее вносят в базу данных по EST. Для поиска гомологии EST с известными генами или семействами генов и для определения категории, к которой относится функция представляемого ей гена, проводят дополнительные сравнения. К 1995 г. было идентифицировано примерно 300 000 EST из 300 кДНК-библиотек 37 органов и тканей. Примерно 90 000 EST представляют собой различающиеся экспрессирующиеся последовательности человека, из них примерно 10 000 соответствуют генам, роль которых в клетке известна, а остальные 80 000 – еще неоткрытым генам.
КЛОНИРОВАНИЕ ГЕНОВ ЗАБОЛЕВАНИЙ ЧЕЛОВЕКА
Как правило, ген конкретного заболевания человека нельзя клонировать, руководствуясь каким-то заранее составленным набором экспериментальных протоколов. Выбор имеющихся в распоряжении исследователя методов и средств зависит от конкретных условий. Начало поиска гена заболевания определяется имеющейся информацией о продукте данного гена. В одних случаях генный продукт бывает хорошо известен, в других можно лишь догадываться, что он собой представляет. Наконец, для многих наследственных заболеваний природа генного продукта вообще неизвестна. Для каждого из этих случаев разработана своя стратегия. В целом для поиска гена заболевания существует четыре подхода: функциональное, кандидатное, позиционное и позииионно-кандидатное картирование. Независимо от применяемого подхода утверждать, что данный ген ассоциирован с интересующим исследователя заболеванием, можно лишь после того, как у больных обнаружены нуклеотидные изменения в гене, не встречающиеся в том же гене у здоровых индивидов.
Выявление мутаций в генах человека
Для выявления мутаций разработан целый ряд простых и недорогих подходов, таких как анализ конформационного полиморфизма одноцепочечной ДНК (SSCP, single-strand conformational polymorphism), градиентный гель-электрофорез в денатурирующих условиях (DGGE, denaturing gradient gel electrophoresis), гетеродуплексный анализ (НА, heteroduplex analysis), химическое расщепление некомплементарных сайтов (CMC, chemical mismatch cleavage), тест на укороченный белок (РТТ, protein truncation test).
Наиболее широко среди перечисленных подходов применяется SSCP. Суть метода состоит в следующем. Как можно большее (по возможности все) число экзонов исследуемого гена по отдельности амплифицируют методом ПЦР, используя в качестве матрицы ДНК больных и здоровых индивидов. Каждая пара праймеров выбирается из последовательностей, фланкирующих экзон, или из его концевых участков. Кроме того, используя данные секвенирования, выбирают праймеры для амплификаиии 5'-области, предшествующей первому экзону гена, 3'-области, следующей за последним экзоном, и участков, содержащих сайты сплайсинга.
ПЦР-продукты каждой реакции денатурируют, быстро охлаждают и разделяют с помощью электрофореза. Благодаря внутри цепочечному спариванию комплементарных оснований и образованию других связей денатурированная одноцепочечная молекула ДНК принимает определенную трехмерную конформацию, зависящую от ее нуклеотидной последовательности. Вследствие комплементарности две цепи одной молекулы ДНК имеют разную нуклеотидную последовательность, а поэтому принимают разную трехмерную конформацию и мигрируют при гель-электрофорезе с разной скоростью. В результате после разделения в геле наблюдаются две полосы, отвечающие разным комплементарным цепям. Если две молекулы ДНК, представляющие один и тот же участок гена, но полученные из разных источников, различаются одной парой нуклеотидов, то с большой вероятностью конформации одиночных цепей таких молекул ДНК будут различаться. Другими словами, каждая из четырех цепей будет перемещаться при гель-электрофорезе со своей скоростью (рис. 20.22). С помощью метода SSCP можно лишь локализовать нуклеотидные изменения в определенном экзоне или специфической области гена, но не определить природу мутации; такую информацию может дать лишь секвенирование Метод SSCP имеет свои ограничения: он выявляет около 90% однонуклеотидных изменений в ПЦР-продуктах длиной не более 200 п. н.
Рис. 20.22. Анализ конформационного полиморфизма одноцепочечной ДНК (SSCP). Препараты ДНК, различающихся одной парой нуклеотидов (А:Т ↔ G:C), амплифицируют ПЦР-методом с использованием одинаковых праймеров (P1, P2). ПЦР-продукты денатурируют и разделяют с помощью гель-электрофореза на двух дорожках (1, 2). Расстояние, на которое перемешается одноцепочечная молекула ДНК, зависит от ее конформации, а последняя, в свою очередь, – от нуклеотидной последовательности. Даже если ДНК различаются лишь одним нуклеотидным сайтом, одиночные цепи могут иметь разную конформацию, а следовательно, ПЦР-продукты образуют в геле не две, а четыре полосы.
Функциональное картирование
Функциональное картирование гена начинается с определения аминокислотной последовательности белка с известной функцией, что позволяет реконструировать нуклеотидную последовательность кодирующей области соответствующего гена (гена-мишени). Основываясь на этих данных, синтезируют олигонуклеотидные зонды и проводят скрининг кДНК-библиотеки, полученной для ткани, в которой данный белок присутствует в большом количестве. Если можно получить очищенную мРНК, с которой транслируется данный белок, то на ней как на матрице можно синтезировать полноразмерную кДНК и клонировать ее. Правильность выбора или синтеза кДНК-клона проверяют секвенированием.
Хромосомную локализацию гена-мишени определяют методом гибридизации in situ с кДНК-клоном или выбранным с его помощью геномным клоном. Для более точной локализациигена-мишени можно также провести скрининг панели монохромосомных клеточных гибридов, а затем и соответствующей делеционной панели при помощи кДНК-клона или отобранного геномного клона.
Затем для определения клонов, гибридизующихся с данным кДНК-клоном, проводят скрининг космидного контига, охватывающего хромосомный район, в котором локализован ген-мишень. Отобранные геномные клоны секвенируют й, используя данные о нуклеотидной последовательности кДНК, идентифицируют экзоны, интроны и 5'-, 3'-фланкирующие последовательности гена. В отсутствие космидного контига, охватывающего район нужной хромосомы, который содержит ген-мишень, выделяют клон с крупной вставкой, содержащей данный район, при помощи кДНК- или геномного зонда. Из клона с крупной вставкой получают субклоны с небольшими вставками, и проводят их скрининг при помощи кДНК-зонда. Позитивные клоны секвенируют и характеризуют ген-мишень (рис. 20.23).
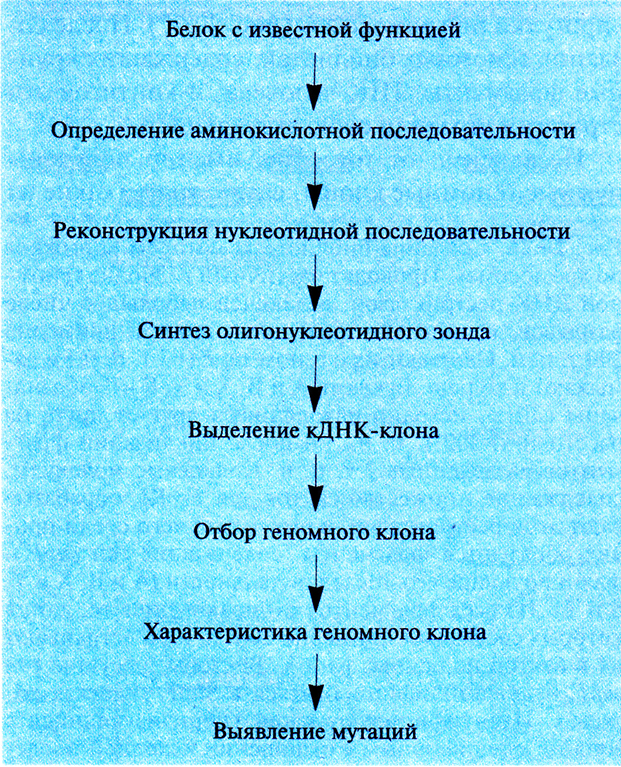
Рис. 20.23. Функциональное картирование Идентификация гена для случая, когда известна аминокислотная последовательность его продукта.
Кандидатное картирование
Хотя этот подход не очень эффективен при картировании генов человека, в ряде случаев он может оказаться весьма полезным. Суть метода состоит в следующем. Анализируют симптомы генетического заболевания и на их основе пытаются понять, какого типа белок может быть с ним ассоциирован. Затем просматривают нуклеотидные последовательности всех клонированных на настоящий момент генов и выбирают ген(ы)-кандидат(ы). Основываясь на нуклеотидной последовательности гена-кандидата, вырабатывают стратегию поиска мутаций и с ее помощью пытаются установить, является ли ген-кандидат искомым геном (рис. 20.24). Принимая во внимание, что геном человека содержит очень большое число генов, а охарактеризованы лишь некоторые из них, не стоит удивляться, что правильный выбор гена случается не так уж часто. Но ненен и отрицательный результат, поскольку он позволяет исключить данный ген из числа ответственных за конкретное генетическое заболевание.
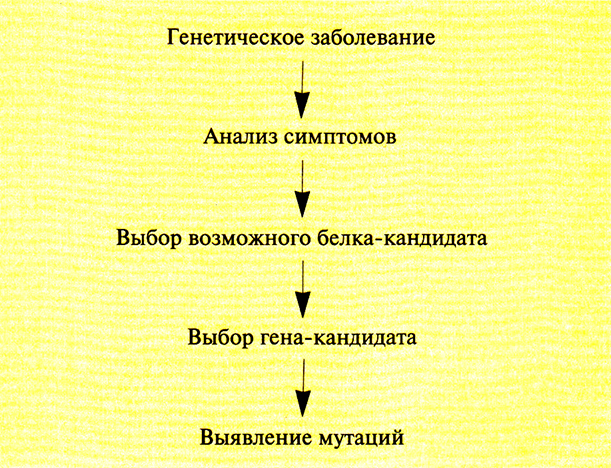
Рис. 20.24. Кандидатное картирование. Идентификация гена, основанная на анализе симптомов обусловленного им заболевания и соображений, какой из уже охарактеризованных генов может претендовать на роль искомого гена.
Позиционное картирование Стратегия позиционного картирования применяется в тех случаях, когда ничего не известно о продукте гена, ответственного за наследственное заболевание, и нет никаких генов-кандидатов (рис. 20.25). В подобных случаях определяют хромосомную локализацию (позицию) гена заболевания и проводят его поиск, применяя различные инструменты и средства («охота за геном»). Благоприятной для позиционного картирования является ситуация, когда у нескольких больных встречается хромосомная перестройка типа транслокации или крупной делеции (> 10 т. п. н.). Предположив, что она затрагивает ген, ответственный за патологический фенотип, при анализе сцепления используют только один специфический район хромосомы вместо того, чтобы проводить сканирование всего генома при помощи большого числа полиморфных маркеров. После локализации гена в конкретном районе хромосомы определяют его положение более точно и идентифицируют ближайшие фланкирующие маркеры, используя мультилокусное картирование с дополнительными полиморфными зондами. Минимальное расстояние между картированными маркерными сайтами, при котором их можно разграничить, в лучшем случае составляет 1 сМ, что соответствует примерно 106 п. н. На таком участке может уместиться в среднем от 20 до 50 генов. Задача позиционного картирования состоит в том, чтобы определить, какой именно из них ответствен за данное заболевание.
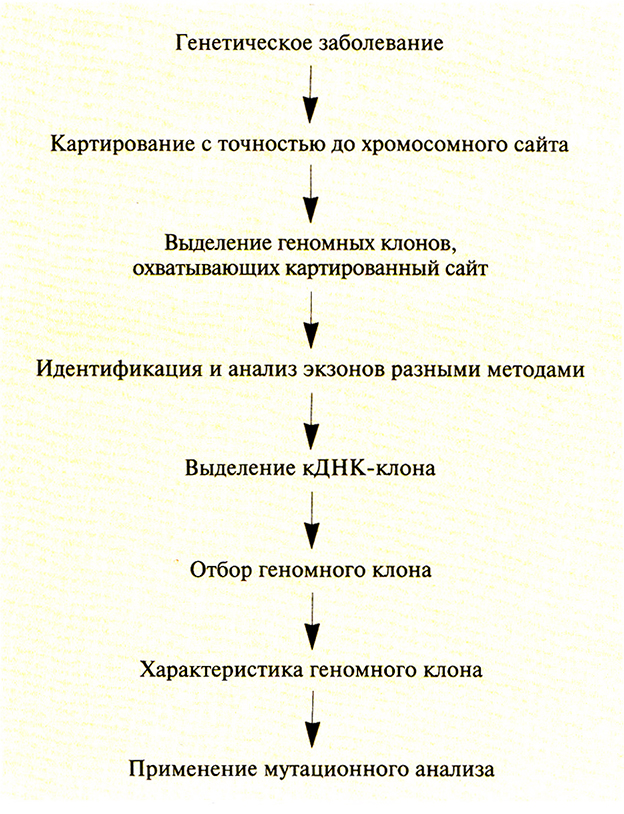
Рис. 20.25. Позиционное картирование. Идентификация гена, продукт которого неизвестен, с помощью хромосомного картирования и зондов, специфичных в отношении тесно сцепленных маркеров.
Из контига, охватывающего район хромосомы, содержащий ген заболевания, выбирают геномные клоны, которые включают фланкирующие маркеры и заключенный между ними участок ДНК. Если такой контиг отсутствует, то с помощью зондов, специфичных в отношении тесно сцепленных маркерных сайтов, проводят скрининг библиотек геномных ДНК для выявления клонов, происходящих из того района, который содержит искомый ген. Между 1986 и 1990 гг., когда метод «охоты за генами» человека еще только разрабатывался, для идентификации нужных геномных клонов использовали метод «прыжков по хромосоме» (рис. 20.26) или метод «прогулки по хромосоме» (рис. 20.27). После создания геномных библиотек, содержащих крупные фрагменты ДНК человека, и контигов эти стратегии утратили свою актуальность.
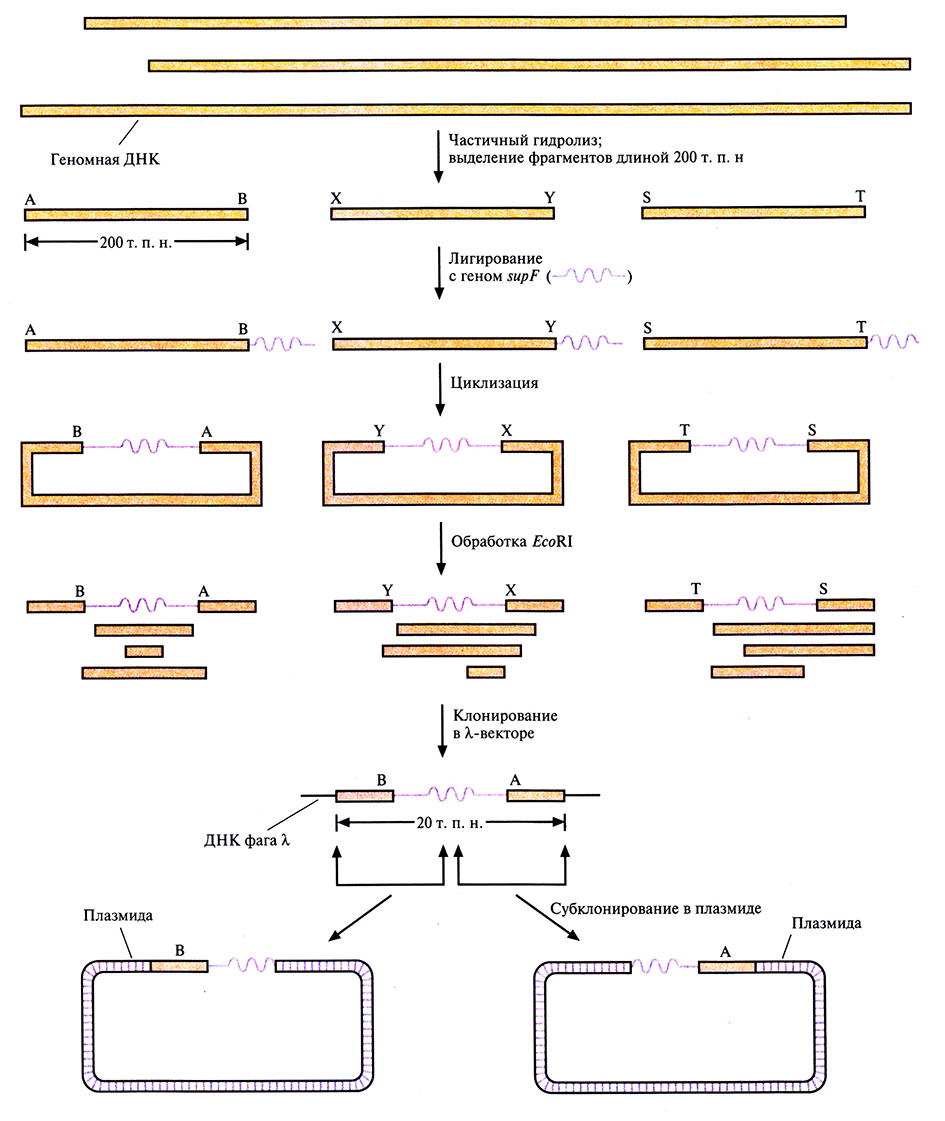
Рис. 20.26. Создание библиотеки методом «прыжков по хромосоме». Проводят частичный гидролиз геномной ДНК рестриктазой, делающей небольшое число разрывов, и выделяют фрагменты длиной примерно 200 т. п. н. Сшивают их с геном supF(~7 т.п.н.) и замыкают в кольцо. Буквами А и В, Х и Y, S и Т обозначены сайты, исходно находящиеся друг от друга на расстоянии 200 т. п. н., но после циклизации фрагментов разделенные 7 т. п. н. Кольцевые молекулы, содержащие множество сайтов для EcoRI, обрабатывают этой рестриктазой, в результате чего среди прочих образуются фрагменты, содержащие ген supF и фланкирующие его последовательности (А и В; X и Y; S и Т). Из всех фрагментов отбирают лишь те, длина которых составляет примерно 20 т.п.н., и встраивают их в вектор на основе фага λ. Векторы, несущие ген supF, будут амплифицироваться в SupF–-клетках-хозяевах. Идентифицируют клон, гибридизуюшийся с зондом, специфичным в отношении исходной последовательности (А, X, S), затем субклонируют его; при этом та его часть, которая не гибридизуется с зондом, содержит участок ДНК (В, Y, Т), находящийся на расстоянии 200 т. п. н. от исходной последовательности.
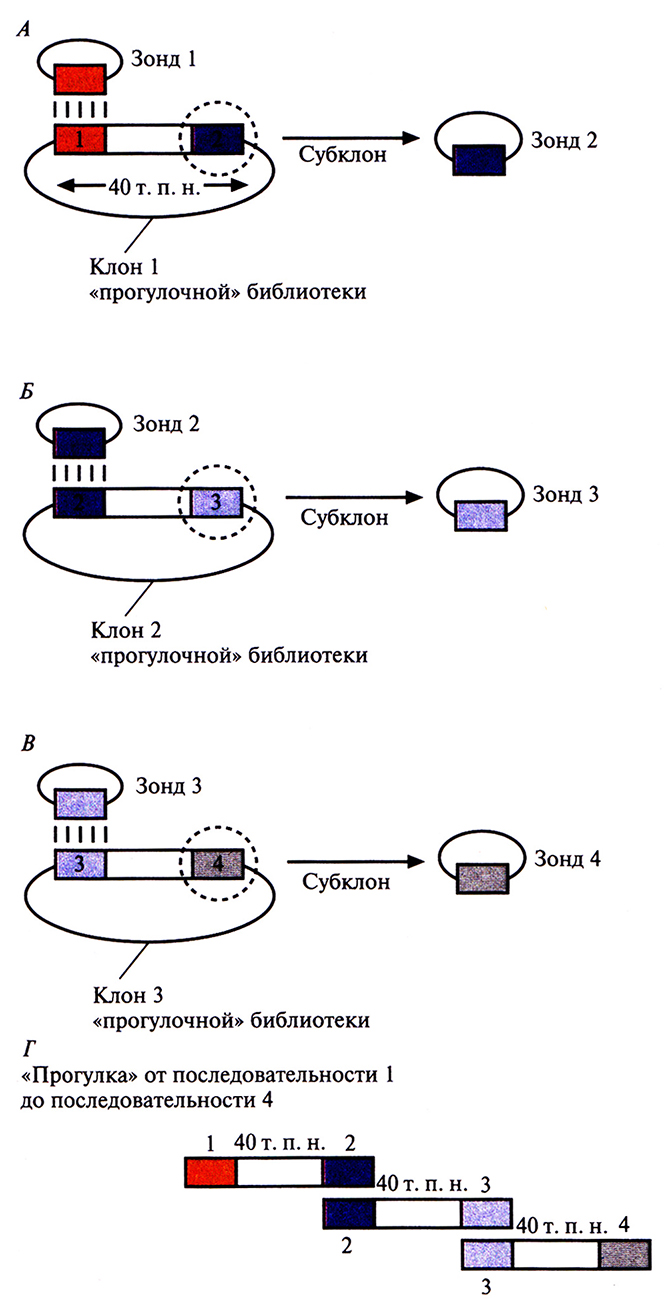
Рис. 20.27. «Прогулка по хромосоме». А. Зонд 1 гибридизуют с клонированным фрагментом ДНК длиной 40 т.п.н. После субклонирования и построения рестрикционной карты последовательность, дистальную по отношению к гибридизовавшейся, используют для создания зонда 2. Б. При помощи зонда 2 из библиотеки выбирают другой клон (отличный от клона 1) и используют последовательность, дистальную по отношению к гибридизовавшейся с ним, для создания зонда 3. Клоны 1 и 2 вместе составляют примерно 80 т. п. н. (за вычетом перекрывающегося участка – зонд 2 – между ними). В. Проводят манипуляции, аналогичные А и Б, используя зонд 3. Третий клон «прогулочной» библиотеки позволяет продвинуться по хромосоме еще на 40 т.п.н. Г. Три перекрывающихся фрагмента ДНК охватывают примерно 120 т.п.н. хромосомной ДНК. «Прогулку» по хромосоме можно совершать в двух направлениях, руководствуясь при этом рестрикционной картой.
Независимо от того, как именно получены нужные геномные клоны, важно знать, какие из них или из субклонов содержат экзоны. Для этого можно использовать целый ряд прямых и косвенных методов, таких как идентификация CpG-островков, межвидовой Саузерн-блоттинг, отбор гибридов, улавливание экзонов, секвенирование ДНК, компьютерный поиск.
Транскрибируемым участкам геномов позвоночных часто предшествуют кластеры нуклеотидов, богатые остатками С и G (CpG-островки). Группу CpG-островков можно идентифицировать по скоплению на рестрикционной карте сайтов для рестрицирующих эндонуклеаз EagI, BssII и SacII. Если как минимум два таких сайта отделены 5–10 т. п. н. друг от друга, значит, они находятся в пределах CpG-островка. Это не гарантирует, что именно здесь находится экзон, но указывает на наличие где-то поблизости транскрибируемого гена.
Геномные клоны или субклоны можно гибридизовать по Саузерну с рестрицированной геномной ДНК различных позвоночных, например с ДНК мыши, крысы, кролика, обезьяны, коровы, цыпленка, рыбы (зооблот, межвидовой блоттинг, блоттинг «Ноев ковчег»). Положительная перекрестная гибридизация означает, что данный клон с большой вероятностью содержит кодирующие последовательности, поскольку многие экзоны в ходе эволюции не изменялись, в то время как повторяющиеся и некодирующие последовательности ДНК, в том числе и интроны, претерпели существенные изменения. Положительный зооблот означает, что клон содержит экзон(ы), однако не показывает, есть ли в нем ген искомого заболевания.
Отбор гибридов позволяет быстро и с высокой эффективностью идентифицировать геномный клон, содержащий экзон(ы), и одновременно изолировать соответствующую кДНК. Отбор можно проводить разными способами. Обычно ДНК геномного клона из той области хромосомы, которая содержит ген заболевания, фиксируют на твердой подложке, проводят прегибридизацию с повторяющимися последовательностями ДНК, а затем гибридизуют с линейными векторными молекулами со вставками из кДНК-библиотеки, происходящей из ткани, вероятнее всего экспрессирующей ген-мишень. Негибридизовавшиеся векторные молекулы смывают с фильтра, а гибридизовавшиеся элюируют и амплифицируют методом ПЦР, используя праймеры из векторных последовательностей, фланкирующих кДНК-вставку (рис. 20.28). Если точно неизвестно, в какой ткани экспрессируется ген-мишень, то кДНК-библиотеки разных тканей объединяют и проводят гибридизацию с отдельными геномными клонами. ПЦР-продукт можно затем клонировать и тестировать, с тем чтобы проверить, содержит ли он кодирующую часть гена данного заболевания. Для этого можно секвенировать кДНК и провести компьютерное сравнение нуклеотидных последовательностей этой ДНК и известных генов. Если будет получена высокая степень гомологии, можно сделать определенные выводы о том, какого типа белок кодирует данная кДНК, и если этот белок таков, что его с высокой вероятностью можно считать продуктом гена-мишени, то данный(е) клон(ы) секвенируют и идентифицируют экзоны, интроны и 5'-, 3'-фланкирующие области. Альтернативный подход состоит в поиске мутаций с целью выявления нуклеотидных различий между ДНКбольных и здоровых индивидов. Если подход, основанный на определении степени гомологии нуклеотидных последовательностей, оказывается безуспешным, то секвенируют и анализируют другие гены из данной области. Реально при поиске гена-мишени для экономии времени и средств характеризуют в первом приближении сразу несколько генов, пока не найдут наиболее вероятный ген-кандидат, который исследуют детально, в том числе с помощью мутационного анализа.
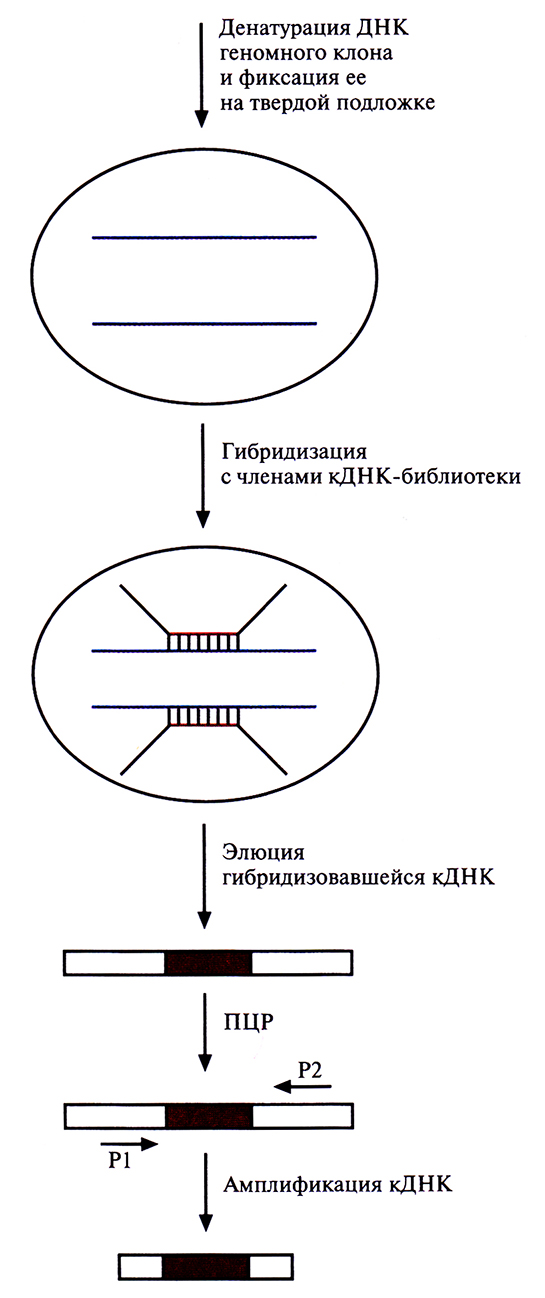
Рис. 20.28. Отбор гибридов. Гибридизацией с ДНК геномного клона «отлавливают» кДНК-клон, амплифицируют его, клонируют и тестируют.
Улавливание экзонов («поимка» экзонов, экзонная амплификация) – это метод, позволяющий идентифицировать и клонировать экзоны, находящиеся в субклонах, полученных из геномных клонов (рис. 20.29). Его суть состоит в следующем. ДНК геномного клона расщепляют так, чтобы получить фрагменты длиной 1–6 т.п.н., и клонируют эти фрагменты в специально сконструированном векторе. Сайт множественного клонирования (полилинкер) вектора расположен внутри интрона, фланкированного двумя экзонами (экзон 1 и экзон 2). Этот искусственный ген (экзон 1–интрон–экзон 2) находится под контролем сильного эукариотического промотора и может реплицироваться в Е. coli или в культуре клеток млекопитающих. После введения (трансфекции) вектора без вставки в клетку млекопитающего происходит транскрипция искусственного гена и удаление интрона из первичного транскрипта. Процессированную РНК (экзон 1–экзон 2) можно выявить, проведя ПЦР-амплификапию обратного транскрипта. Сначала с помощью обратной транскриптазы на мРНК синтезируют ДНК (синтез первой цепи). Вторую цепь синтезируют при участии праймера, комплементарного части экзона 1 первой цепи. Затем в реакционную смесь добавляют второй праймер, комплементарный части экзона 2 второй цепи ДНК, и проводят ПЦР-амплификацию. Длину ПЦР-продукта определяют с помощью гель-электрофореза.
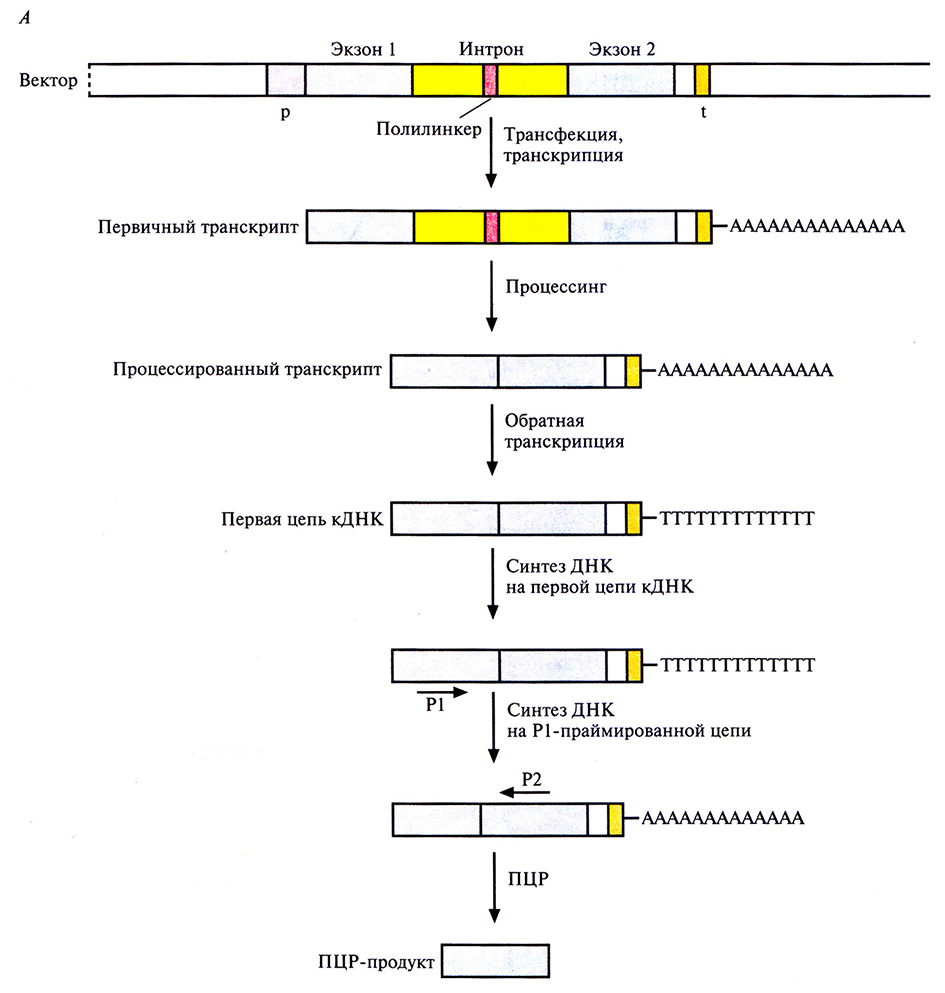
Рис. 20.29. Улавливание экзонов. А. Вектор для улавливания экзонов содержит искусственный ген, состоящий из промотора р, двух экзонов, разделенных интроном, который несет полилинкер, и сайта терминании транскрипции t. После введения вектора в эукариотическую клетку искусственный ген транскрибируется и из первичного транскрипта удаляется интрон. Для получения ПЦР-продукта определенной длины, который содержит часть обоих экзонов, используют ПЦР-амплификацию обратного транскрипта.
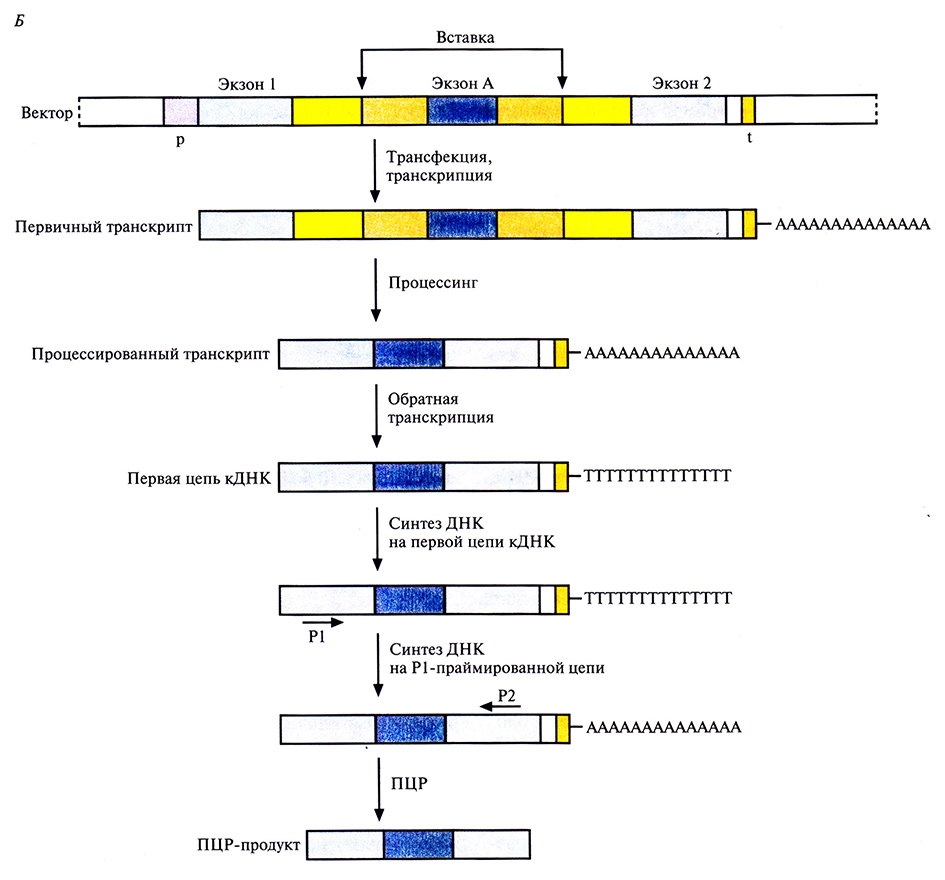
Рис. 20.29. Б. В полилинкер вектора для улавливания экзонов встроен фрагмент ДНК человека, и эта конструкция введена в эукариотическую клетку (трансфекция). В данном случае экзон содержит функциональные акцепторный и донорный сайты сплайсинга. При процессинге первичного транскрипта удаляются интроны, фланкирующие экзон А, и он оказывается между экзонами 1 и 2 Длина ПЦР-продукта обратного транскрипта показывает, «пойман» ли искомый экзон между экзонами 1 и 2 и, следовательно, содержит ли его данная вставка.
Если в вектор встроить рестрикционный фрагмент, содержащий некий экзон А и фланкирующие его интроны, то после трансфекции процессированный транскрипт будет содержать три экзона: экзон 1–экзон А–экзон 2. Длина ПЦР-продукта будет больше, чем в тех случаях, когда в векторе нет вставки, когда вставка содержит экзон без функциональных сайтов сплайсинга (донорного и акцепторного) или когда вставка вообще не содержит экзона. Если во вставке присутствует более одного экзона, каждый из которых имеет функциональные сайты сплайсинга, то процессированный транскрипт будет содержать все эти экзоны.
В том случае, если в каждом праймере содержатся рестрикционные сайты, клонируют ПЦР-продукт, несущий «пойманный» экзон, и используют последний в качестве зонда для скрининга кДНК-библиотеки. Зная нуклеотидную последовательность «пойманного» экзона, предпринимают поиск гомологичных ему последовательностей в базе данных. Если есть основания полагать, что «пойманный» экзон с большой вероятностью является частью гена данного заболевания, то характеризуют и секвенируют геномные клоны, охватывающие место расположения данного гена, и исследуют образцы ДНК больных и здоровых индивидов с целью выявления мутаций. Поскольку мутации, ответственные за патологию, не всегда бывают равномерно распределены по всем экзонам, чем больше размер сканированной кодирующей области предполагаемого гена, тем больше вероятность обнаружения мутации.
Для идентификации экзонов используют различные компьютерные программы, например GRAIL (Gene Recognition and Analysis Internet Line). Они созданы исходя из некоторых характерных для экзона особенностей. Одна из них – ожидаемая нуклеотидная последовательность кодирующей области. Если лаборатория оснащена оборудованием для широкомасштабного секвенирования, можно секвенировать геномные клоны, охватывающие область расположения искомого гена, и провести компьютерную обработку полученных данных с целью выявления экзонов. Нуклеотидную последовательность предполагаемого экзона можно использовать для поиска гомологичных ей последовательностей в генной базе данных или синтезировать на ее основе олигонуклеотидный зонд для скрининга кДНК-библиотеки. Наконец, как и в случае других методов идентификации экзонов в геномных клонах, необходимо доказать, что предполагаемый экзон является частью гена-мишени.
Реализация любого проекта по позиционному картированию гена занимает много времени. За период с 1986 по 1995 г. с помощью данного подхода удалось обнаружить более 50 генов различных заболеваний человека, что можно считать большим достижением. Иногда поиск гена занимает 1–2 года, в то же время для обнаружения гена хореи Гентингтона консорциуму из нескольких исследовательских лабораторий потребовалось 10 лет. Отметим, что с клонированием все новых и новых генов и построением транскрипционных карт с высоким разрешением позиционное картирование постепенно уступает место позиционно-кандидатному.
Позиционно-кандидатное картирование
Позиционно-кандидатное картирование состоит в определении хромосомной локализации гена болезни, продукт которого неизвестен, и последующем анализе современных генетических и транскрипционных карт, с тем чтобы выявить кодирующие последовательности (гены, внутригенные EST), находящиеся в этом же районе (рис. 20.30). Весьма вероятно, что одна из этих последовательностей и окажется геном данного заболевания. Если какой-либо из генов-кандидатов охарактеризован, можно провести его мутационный анализ. Как альтернативу можно использовать «кандидатные» EST в качестве зондов, отобрать с их помощью геномный клон и секвенировать его, а затем также провести мутационный анализ. По мере детализации физических и транскрипционных карт позиционно-кандидатное картирование становится все более популярным при поиске генов различных заболеваний человека.
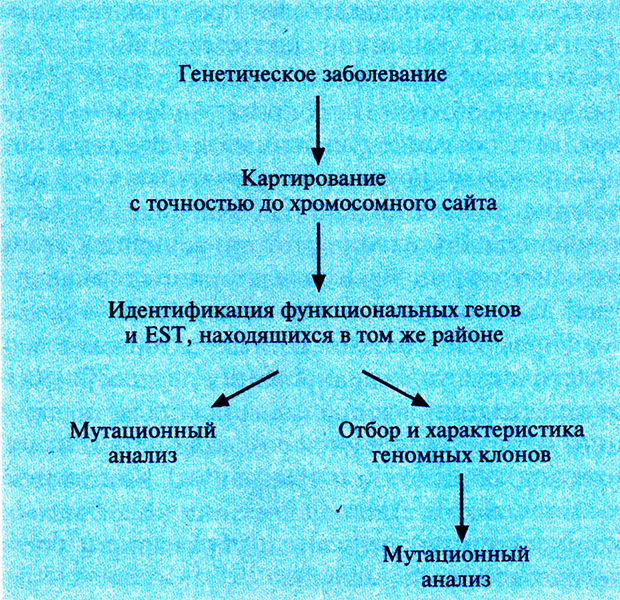
Рис. 20.30. Позиционно-кандидатное картирование. Идентификация гена заболевания в том случае, когда продукт гена неизвестен, но ген картирван в том же хромосомном районе, что и некоторые функциональные гены и EST. Из этих генов и EST отбирают «кандидатные» и определяют, какие из них соответствуют искомому гену.
ПРОГРАММА «ГЕНОМ ЧЕЛОВЕКА»
Работа над реализацией программы «Геном человека» (HGP, Human Genome Project) официально началась 1 октября 1990 г. в США и контролнру ет ее Министерство энергетики (Department of Energy) совместно с Государственными институтами здоровья (National Institutes of Health) США. Ее конечная цель состоит в определении нуклеотидной последовательности всего генома человека. Полученная обширная генетическая информация станет основой для более узких проектов исследования всех моногенных генетических заболеваний и послужит трамплином для изучения сложных наследственных патологий. В 1990 г. предполагалось, что работа в рамках HGP займет 15 лет, а ее стоимость составит 2 млрд. долл. За короткое время программа стала международной, ее проекты финансируются правительствами Великобритании, Франции. Канады, Германии и Японии. В настоящее время происходит кооперация и координация усилий многих государственных и межгосударственных агентств, частных компаний и некоммерческих исследовательских институтов. HGP – обширная программа, охватывающая множество различных направлений.
Та часть программы «Геном человека», которая выполняется в США, включает следующие подпрограммы: построение генетических и физических карт с высоким разрешением; снижение себестоимости и повышение эффективности крупномасштабного секвенирования ДНК; разработка новых технологий картирования генов и секвенирования ДНК; усовершенствование компьютерных технологий для обработки и хранения больших массивов данных; изучение этического, правового и социального аспектов исследований, проводимых в рамках программы. Цель последней подпрограммы состоит в создани руководств для исследователей и врачей и обосновании политики правительства, касающейся использования генетической информации. В рамках подпрограммы «Новые технологии» консорциумом нескольких групп исследователей был полностью секвенирован геном дрожжей Saccharomyces сегеvisiae (150 м.п.н.). Кроме того, определены полные нуклеотидные последовательности ДНК других «модельных» организмов, таких как нематоды (Caenorhabditis elegans, 100 м. п. н.), плодовая мушка (Drosophila melanogaster, 120 м. п. н.), бактерии (С. coli. 4,2 т. п. н.), мышь (Mus musculus, 3000 м.п.н.).
Некоторые из намеченных на период 1990–1995 гг. задач той части HGP, которая выполняется в США, в 1993 г. были пересмотрены; это связано с быстрым прогрессом в генетическом картировании благодаря внедрению микросателлитных полиморфных маркеров и построению практически полных физических карт. К 1996 г. удалось решить несколько вновь поставленных задач. Например, в 1994 г. была опубликована карта (генетического) сцепления человека, которая содержала 5826 локусов, охватывающих 4000 сМ. Хотя только для 908 локусов шансы сцепления составили больше 1000:1, конечная плотность маркеров равнялась 0,7 сМ (4000 сМ/5826 локусов), что больше ожидаемой в 1995 г. плотности карты 2–5 сМ. К 1996 г. было запланировано построение физической карты, основанной на STS, с разрешением 300 т. п. н., то есть один STS-сайт должен был приходиться на каждые 300 т.п.н. ДНК человека. Однако уже в 1995 г. была построена полная физическая карта с разрешением 200 т.п.н.
Успехи в оптимизации технологии секвенирования ДНК вполне ощутимы, но не столь эффектны. Стоимость секвенирования пары оснований снизилась с 5 долл. США в 1990 г. до 0,3 долл. в 1996 г. Скорость секвенирования возросла с 10 000 оснований в день в 1990 г. до 50 000 в 1996 г. К 1998 г. предполагается секвенировать 80 м. п. н., или 2,5%, генома человека. Если не произойдет никаких кардинальных изменений, то при помощи «фабрики» из 30 автоматических секвенаторов, работающих круглые сутки, и полного набора физических карт космидных клонов можно будет секвенировать примерно 3000 м.п.н. ДНК за 6 лет, потратив на это -900 млн. долл. США. Какие-то время и средства придется потратить еще на проверку ошибок и получение окончательной последовательности. Однако, прежде чем приступать к реализации столь крупномасштабного проекта, ученые пытаются добиться значительного повышения скорости секвенирования при помощи автоматических флуоресцентных секвенаторов, в которых используется метод Сэнгера. Кроме того, предпринимаются поиски других способов быстрого секвенирования ДНК.
Чтобы регулировать работу над различными аспектами всей программы, HGP распределяет финансы между разными исследовательскими группами. В большинстве случаев ответственность за создание генетических и физических карт конкретной хромосомы делят между собой крупные центры и небольшие лаборатории, которые сотрудничают друг с другом. Некоторые из наиболее крупных исследовательских институтов занимаются укомплектованием данных о генетических и физических картах генома. В результате молекулярно-генетических исследований генома человека появляется огромное количество новых данных о полиморфных зондах, STS-клонах, содержании генетических, физических и объединенных карт, рестрикционных фрагментах, геномной дактилоскопии и нуклеотидных последовательностях ДНК. Эти данные необходимо собирать, упорядочивать, хранить, объяснять, сравнивать, объединять и предоставлять другим исследователям как в исходном, так и в окончательном виде. Эффективное использование этой информации было бы невозможно без компьютерного обеспечения, включающего в себя базы данных, системы управления базами данных, алгоритмы математического моделирования и программы автоматизации экспериментов. Область знаний, которая занимается созданием численных методов обработки информации, называется информатикой. Биоинформатика имеет дело с компьютерным анализом и управлением биологической информацией.
В рамках программы HGP, касающейся усовершенствования компьютерных технологий, достигнуты значительные успехи в создании компьютерных программ, позволяющих проводить всестороннюю обработку данных по геному человека. Созданы электронные сайты, где специалисты и общественность могут получить информацию о содержании различных хромосомных карт, включая их полное графическое изображение, о методах исследования генома и программном обеспечении. Например, WWW-сайт Государственного центра по изучению генома человека в США (http://www.nhgri.nih.gov/index.html) содержит информацию о программе «Геном человека» и множество ссылок на другие центры, занимающиеся этой проблемой.
С самого начала своего существования HGP должна была решать этические, правовые и социальные проблемы, связанные с картированием и секвенированием генома человека, вырабатывать стратегию, тактику и разрабатывать законопроекты, гарантирующие ответственное использование информации по генетике человека. На самом деле HGP не ставит каких-либо принципиально новых этических, правовых или социальных вопросов, которые не возникали бы при проведении медико-генетических исследований в целом. Однако реализация HGP неизбежно приведет к идентификации большого числа генов различных заболеваний и к определению последовательности многих из них, и эта информация будет использоваться при разработке ДНК-диагностических тестов.
Здесь возникает множество поводов для беспокойства. Не будет ли генетическая информация использоваться для дискриминации людей при медицинском страховании, приеме на работу или иммиграции? Не приведет ли ее доступность к социальному неравенству? Все ли меры приняты, чтобы сохранить конфиденциальность персональной генетической информации? Как найти баланс между нуждами личности и общества? Обладают ли частнопрактикующие врачи и врачи, работающие в клиниках, достаточными знаниями по медицинской генетике, чтобы они могли разъяснить пациентам смысл конкретного генетического теста? Сможет ли генетическое консультирование уменьшить обеспокоенность обратившегося? Не скажется ли отрицательно доступность генетической информации на семейных отношениях? Можно ли надеяться на то, что удастся получить согласие на проведение диагностического теста у достаточно осведомленного пациента? Следует ли предлагать тестирование в том случае, когда данное наследственное заболевание неизлечимо? Как повысить образовательный уровень населения, чтобы оно понимало значение генетической информации? На эти и многие другие вопросы, возникающие при изучении генетики человека, нет однозначных ответов. В США в рамках подпрограммы по изучению этического, правового и социального аспектов генетических исследований организован целый рад мероприятий: разработаны обучающие программы, проводятся семинары и выставки для студентов, учителей, врачей, общественности, адвокатов и судей; исследована возможность генетического тестирования муковисцидоза и наследственных форм рака молочной железы, яичников и толстой кишки, созданы две комиссии (по генетической информации и страхованию; по генетическому тестированию) для исчерпывающего изучения конкретных вопросов; разрабатываются предложения для выработки федеральных законов США, которые обеспечивали бы конфиденциальность генетической информации, получаемой при идентификации личности.
На основе этой программы и других исследований были сформулированы пять основных принципов, которыми следует руководствоваться при использовании генетической информации и в работе генетических консультаций: право на автономию, конфиденциальность, справедливость, беспристрастность и качество. Концепция права на автономию в данном случае означает необходимость соблюдения прав человека, обращающегося в генетическую консультацию. Например, генетическое тестирование должно проводиться добровольно и только после того, как пациент в достаточной степени информирован; тестированию должны подвергаться лишь лица, относящиеся к группе риска; тестируемые должны сами решать, будут ли они знакомиться с результатами теста. Консультируемые должны быть хорошо осведомлены о всех особенностях теста: его прогностической ценности, медицинских аспектах, характере терапии, если она возможна.
Обычно считается, что генетическая информация отличается от других видов личностной информации, поэтому необходимо предусмотреть особые меры предосторожности, гарантирующие ее конфиденциальность. Справедливость и беспристрастность – это довольно близкие понятия. Достигнуто соглашение, что генетическое консультирование должно быть доступно всем, кто в нем нуждается. Как и в случае социальных и медицинских программ, необходимо защитить права умственно неполноценных пациентов и детей. Что касается качества, то тестирование должны проводить высококвалифицированные сотрудники, используя при этом надлежащие методы и средства; все этапы должны соответствующим образом контролироваться, чтобы гарантировать правильность их использования.
ЗАКЛЮЧЕНИЕ
Изучая родословные семей, представленных несколькими поколениями, члены которых имеют четко выраженную патологию, можно определить тип наследования многих генетических заболеваний. Зная характер наследования в семьях, можно установить, является ли данное генетическое заболевание аутосомно-доминантным, аутосомно-рецессивным, Х-сцепленным доминантным или Х-сцепленным рецессивным. В случае Х-сцепленного заболевания его ген расположен на Х-хромосоме, для аутосомных болезней хромосомная локализация гена неизвестна. Чтобы картировать ген в специфическом районе хромосомы, можно идентифицировать сцепленные с ним маркерные сайты, используя для этого метод ПДРФ и STRP-картирование. Для опенки сцепления между маркерным сайтом и геном заболевания используют метод максимального правдоподобия. Порядок расположения ПДРФ- и STRP-сайтов на хромосоме определяют при помощи анализа наследования гаплотипов в группе семей, представленных тремя поколениями и имеющих большое количество детей (СЕРН-семей). Кроме того, порядок расположения на хромосоме уникальных сайтов, идентифицируемых при помощи ПЦР (STS), можно проверить картированием с использованием радиационных гибридов. Физические карты хромосом (контиги) строят на основе геномных библиотек, содержащих крупные (YAC, ВАС и РАС) и небольшие (космиды, Р1 и λ) фрагменты ДНК человека, используя STS-картирование или другие подходы, в том числе геномную дактилоскопию. Транскрипционные карты состоят из участков кДНК и маркерных экспрессируемых последовательностей (EST), расположенных вдоль хромосомы. Построение генетических, физических и транскрипционных карт облегчает идентификацию и характеристику генов заболеваний.
Аутентичность обнаруженного гена человека можно считать доказанной, если у больных индивидов в нем найдены изменения, отсутствующие в генах здоровых лиц. Для выявления мутаций часто используют анализ конформационного полиморфизма одноцепочечной ДНК (SSCP).
Для идентификации нужного гена человека используют четыре метода. В первом из них, функциональном картировании, на основе данных о генном продукте синтезируют зонды для скрининга кДНК-библиотеки. Положительный кДНК-клон, содержащий кодирующую область гена-мишени, используют для отбора геномных клонов и характеристики гена в целом. Второй подход, кандидатное картирование, основывается на выборе генов, которые по имеющимся данным могут отвечать за данное генетическое заболевание. В этом случае проводят поиск мутаций в генах-кандидатах у больных и здоровых индивидов и по результатам поиска делают вывод, какой из них является геном заболевания. Третий подход, позиционное картирование, применяют в тех случаях, когда ничего не известно ни о возможном гене заболевания, ни о его продукте. Этот подход весьма трудоемок и имеет множество модификаций. Сначала, используя ПДРФ- или STRP-зонды и данные о семьях с исследуемым наследственным заболева нием, определяют район хромосомы, в котором локализован искомый ген. Затем с помощью зондов, специфичных в отношении тесно сцепленных с ним маркеров, выявляют клоны, охватывающие район локализации гена заболевания Проверяют геномные клоны или полученные из них субклоны на наличие в них экзонов. Используя данные о нуклеотидных последовательностях различных экзонов, в той или иной степени соответствующих нуклеотидной последовательности гена заболевания, разрабатывают стратегию поиска мутаций. Четвертый подход, позиционно-кандидатное картирование, состоит в картировании гена заболевания в определенном районе хромосомы, просмотре функциональных генов и маркерных экспрессируемых последовательностей, локализованных в том же хромосомном районе, и выборе тех из них, которые могут являться искомым геном. Чтобы определить, какой именно из генов-кандидатов является таковым на самом деле, используют мутационный анализ.
«Геном человека» – это широкомасштабная исследовательская программа, конечной целью которой является полное секвенирование генома человека. Различные ее направления включают построение генетических и физических карт всех хромосом человека с высоким разрешением; секвенирование геномов различных модельных организмов тина Е. coli, С. elegans, S. сегеvisiae, М. musculus и A. thaliana; создание компьютерных технологий для обработки и анализа данных по генетическому и физическому картированию и секвенированию ДНК; информирование общественности по всем проблемам, связанным с получением и использованием данных по генетике человека, изучение этических, правовых и социальных аспектов генетических исследований. На этом пути уже достигнуты впечатляющие успехи, и есть основания полагать, что геном человека будет секвенирован к 2005 г.
