Технология рекомбинантных ДНК позволяет выделять гены любых белков, существующих в природе, экспрессировать их в специфическом хозяйском организме и получать чистые белковые продукты. Однако физические и химические свойства таких «природных» белков часто не удовлетворяют условиям, обеспечивающим возможность их промышленного применения. Иногда для получения белков, обладающих нужными свойствами, в качестве источника соответствующих генов используют организмы, растущие в необычных, зачастую экстремальных условиях. Например, для синтеза а-амилазы, не утрачивающей своей активности при высокой температуре, выделили ее ген из Bacillus stearothermophilus – бактерии, естественной средой обитания которой являются горячие источники с температурой воды 90 °С. Полученная таким образом α-амилаза оставалась активной при температурах, при которых осуществляют промышленное производство этилового спирта из крахмала. Для получения белков с заранее заданными свойствами можно использовать также мутантные формы генов. Однако число мутантных белков, образующихся в результате замены отдельных нуклеотидов в структурном гене с помощью обычного мутагенеза, чрезвычайно велико. Мутагенез с последующим отбором редко приводит к существенному улучшению свойств исходного белка, поскольку большинство аминокислотных замен сопровождается снижением активности фермента.
Для создания белков со специфическими свойствами можно использовать другой подход, основанный на внесении изменений в кодирующие их клонированные гены. Это позволяет получать белки с другими, чем у их аналогов, свойствами.
- Изменив константу Михаэлиса (KM), которая характеризует прочность связывания субстрата с ферментом, и максимальную скорость (Vmах) превращения субстрата в продукт при определенных условиях, можно повысить общую каталитическую эффективность (Vmах/ KM) реакции; Vmax равна полному количеству фермента (E0), умноженную на каталическую константу (kcat).
- Повысив стабильность белка в широком диапазоне температур или рН, можно использовать его в условиях, при которых исходный белок инактивируется.
- Создав белки, способные функционировать в безводных растворителях, можно осуществлять каталитические реакции в нефизиологических условиях.
- Изменив белок таким образом, чтобы он мог работать без кофактора, можно использовать его в некоторых непрерывных промышленных процессах.
- Изменив активный центр фермента, можно повысить его специфичность и уменьшить число нежелательных побочных реакций.
- Повысив устойчивость белка к клеточным протеазам, можно упростить процедуру его очистки и повысить выход продукта.
- Изменив аллостерическую регуляцию фермента, можно уменьшить степень его ингибирования метаболитом по типу отрицательной обратной связи и увеличить выход продукта.
НАПРАВЛЕННЫЙ МУТАГЕНЕЗ: МЕТОДИКА
Получить новый белок с заранее заданными свойствами – непростая задача, но вполне реально изменить свойства уже существующего белка. Изменения можно вносить в сам белок или в его ген. Однако химическая модификация белков редко бывает строго специфичной и ее необходимо осуществлять заново для каждого белкового препарата, поэтому лучше вносить изменения в его клонированный ген. К сожалению, не всегда бывает известно, какую именно аминокислоту или последовательность аминокислот нужно изменить, чтобы получить белок с нужными физическими, кинетическими или химическими свойствами. Может случиться, что изменения должны затрагивать два или более аминокислотных остатка, расположенных далеко друг от друга в полипептидной цепи, но сближающихся в результате укладки белковой молекулы. Есть надежда, что уже в недалеком будущем с помощью компьютеров удастся предсказывать свойства того или иного белка, исходя из данных о его аминокислотной последовательности. Это значительно упростит процедуру создания нужных белков. Введение новой генетической информации в клонированные гены сейчас не составляет особого труда, однако, чтобы определить, обладает ли искомый белок нужными свойствами, необходимо проанализировать множество белковых продуктов.
Внесение специфических изменений в кодирующие последовательности ДНК, приводящих к определенным изменениям в аминокислотных последовательностях, называется направленным мутагенезом. Идентификация аминокислот, замена которых даст желаемый результат, облегчается, если детально известна пространственная структура белка (ее устанавливают с помощью рентгеноструктурного анализа или других аналитических методов). Однако для большинства белков такие данные отсутствуют, поэтому направленный мутагенез – это в значительной мере эмпирическая процедура, основанная на методе проб и ошибок. Каждый белок, кодируемый мутантным геном, нужно протестировать и убедиться в том, что мутация дала желаемый эффект.
Для направленного мутагенеза клонированных генов используют разные экспериментальные подходы. В одних случаях вносят изменения в специфические сайты клонированного гена, в других случайным образом изменяют короткий фрагмент клонированного гена и среди образующихся мутантных белков выбирают один, обладающий необходимой активностью.
Олигонуклеотид-направленный мутагенез
с использованием ДНК фага М13
Олигонуклеотид-направленный (сайт-специфический) мутагенез – это один из наиболее простых методов внесения точковых мутаций в клонированный ген (рис. 8.1). Для его осуществления необходимо знать: 1) точную нуклеотидную последовательность той области ДНК, которая соответствует мРНК-кодону, подлежащему изменению; 2) характер аминокислотных замен. Обычно встраивают ген-мишень в двухцепочечную форму вектора на основе бактериофага М13. Сначала выделяют одноцепочечную форму вектора (плюс-цепь М13) и смешивают ее с синтетическим олигонуклеотидом, в точности комплементарным – за исключением одного нуклеотида – нужному сегменту клонированного гена. Этот отличающийся (т. е. неспаривающийся) нуклеотид соответствует тому нуклеотиду кодона мРНК, который необходимо изменить. В случае, представленном на рис. 8.1, триплет АТТ, соответствующий изолейциновому кодону AUU, нужно заменить на триплет СТТ, соответствующий лейциновому кодону CUU. Олигонуклеотид будет гибридизоваться с комплементарным участком клонированного гена в том случае, если: 1) он добавлен в количестве, во много раз превышающем количество ДНК М13; 2) неспаривающийся нуклеотид находится примерно посередине олигонуклеотида; 3) отжиг проводят при низкой температуре и высокой ионной силе. 3'-конеп спарившегося олигонуклеотида служит затравкой для инициации синтеза ДНК, а интактная цепь ДНК М13 – матрицей. Репликация осуществляется с помощью фрагмента Кленова ДНК-полимеразы I Escherichia coli при наличии в среде четырех дезоксирибонуклеозидтрифосфатов, а присоединение последнего нуклеотида синтезированной цепи к 5'-концу затравки обеспечивает ДНК-лигаза фага Т4. Однако in vitro синтез ДНК редко идет до конца, и частично двухцепочечные молекулы приходится отделять от нормальных центрифугированием в градиенте сахарозы.
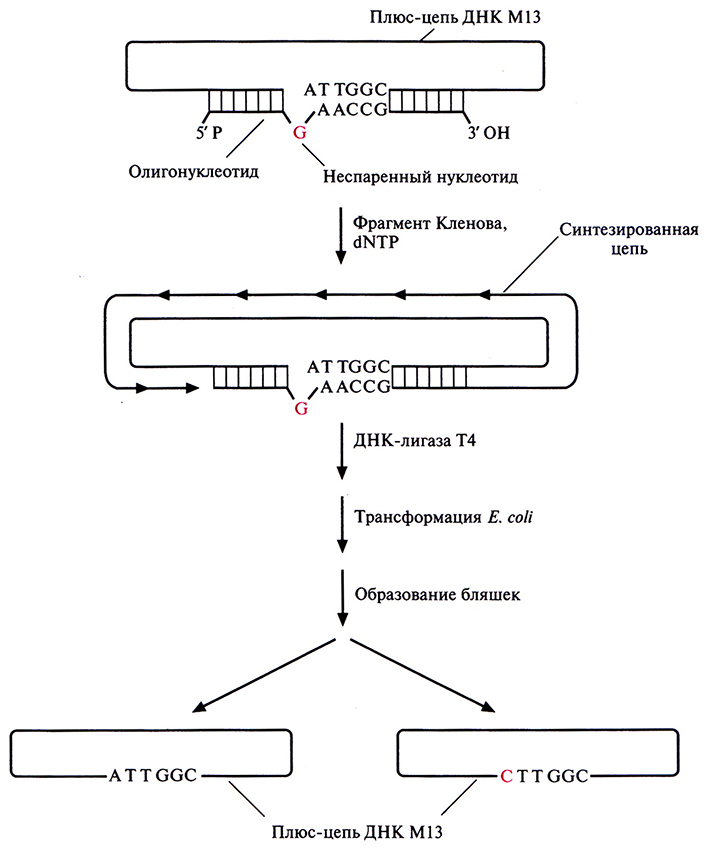
Рис. 8.1. Олигонуклеотид-направленный мутагенез. Одноцепочечную ДНК фага М13 (плюс-цепь), несущую ген-мишень, отжигают с комплементарным синтетическим олигонуклеотидом, содержащим одно основание, не комплементарное соответствующему основанию исходной ДНК. Олигонуклеотид служит затравкой для синтеза ДНК, а М13-вектор с встроенным геном – матрицей. Репликацию катализирует фрагмент Кленова ДНК-полимеразы I Е. coli. Синтезированную полноразмерную цепь замыкает в кольцо ДНК-лигаза Т4. Образовавшимися двухцепочечными молекулами трансформируют Е. coli. Часть фаговых частиц содержит ДНК дикого типа, часть – мутантную ДНК.
Полностью двухцепочечными молекулами ДНК фага М13, содержащими, однако, некомплементарные нуклеотиды, трансформируют клетки Е. coli. В последних образуются фаговые частицы, что в конечном счете приводит к лизису клеток и образованию бляшек. Поскольку репликация идет по полуконсервативному механизму, половина популяции образующихся фаговых частиц должна содержать ДНК дикого типа, а половина – мутантную ДНК со специфической нуклеотидной заменой. Частицы, содержащие только мутантный ген, идентифицируют при помощи ДНК-гибридизации в жестких условиях, используя в качестве зонда исходный олигонуклеотид. Мутантный ген вырезают и встраивают в какой-либо экспрессирующий Е. соli-вектор. Мутантный белок синтезируют в Е. coli и очищают.
На самом деле число фаговых частиц, несущих мутантную ДНК, оказывается гораздо меньше ожидаемых 50%: лишь 1–5% бляшек содержат фаг с мутантным геном. Чтобы повысить выход мутантного фага, метод олигонуклеотид-направленного мутагенеза модифицировали. Один из подходов состоял во введении М13-вектора, несущего ген, в который необходимо внести мутацию, в штамм Е. coli, дефектный по двум ферментам метаболизма ДНК (рис. 8.2). Один фермент – это мутантная форма dUTP-пирофосфатазы (dut). Клетки с неактивной dUTP-пирофосфатазой характеризуются повышенным содержанием dUTP, что приводит квстраиванию в ДНК при репликации нескольких остатков dUTP вместо dTTP. Второй фермент – это дефектная урацил-N-гликозилаза (ung). В отсутствие функциональной урацил-N-гликозилазы остатки dUTP, случайно встроившиеся в ДНК, не могут быть удалены. В одноцепочечной ДНК М13, синтезированной в таких клетках Е. coli, примерно 1% тимидиновых остатков оказываются замененными уридиновыми. Олигонуклеотид с некомплементарным основанием отжигают с урацилсодержащей ДНК М13 и in vitro достраивают вторую цепь. Двухцепочечной ДНК трансформируют штамм Е. coli, содержащий функциональный ген ung. Активная урацил-N-гликозилаза хозяйских клеток удаляет остатки уридина из ДНК М13 (рис. 8.2), исходная матричная цепь М13 деградирует и далее реплицируется только мутантная цепь, не содержащая dUTP. В результате выход фаговых частиц, несущих мутантный ген, значительно увеличивается.
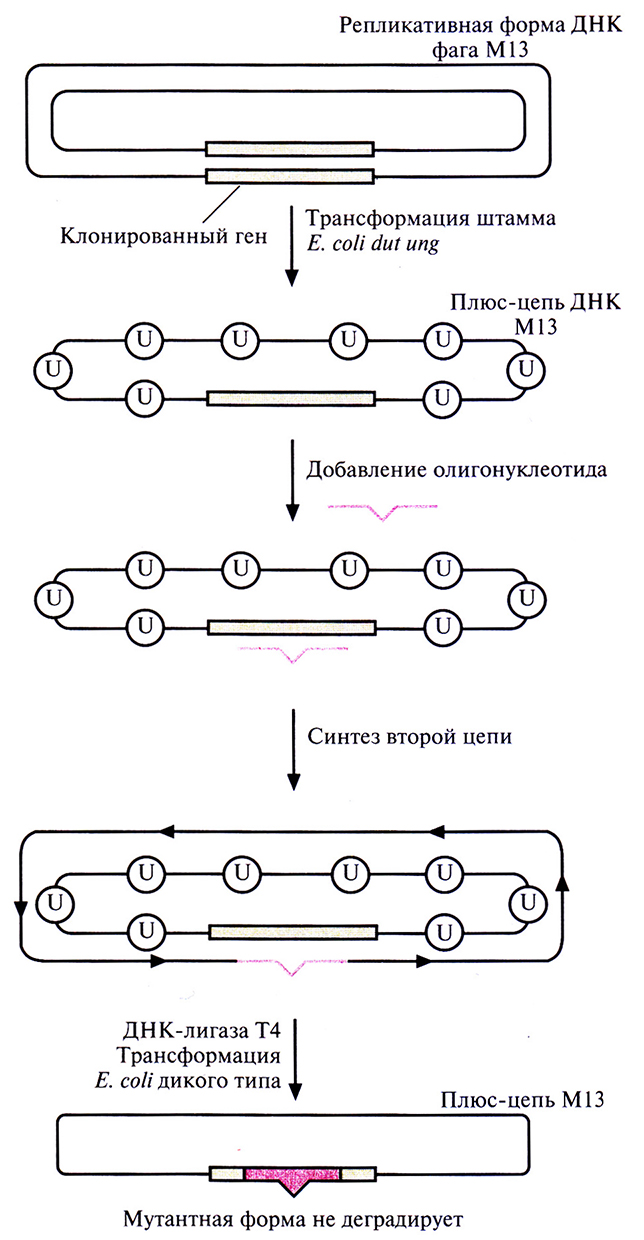
Рис. 8.2. Повышение выхода мутантного фага М13 путем трансформации штамма Е. coli dut ung. Ген-мишень встраивают в двухцепочечную репликативную форму ДНК фага М13 и полученными молекулами трансформируют штамм Е. coli dut ung. Мутация dut вызывает повышение содержания dUTP в клетке, что приводит к включению в ДНК нескольких остатков dUTP (U), а мутация ung блокирует их удаление. Двухцепочечной ДНК М13, содержащей ген-мишень, трансформируют клетки Е. coli дикого типа. Продукт гена ung дикого типа (урацил-N-гликозилаза) удаляет все остатки урацила из исходной цепи, и она деградирует. Мутантная цепь остается интактной, поскольку она не содержит остатков урацила. Эта цепь служит матрицей для репликации ДНК, и в результате доля фаговых частиц, несущих мутантный ген, увеличивается.
Олигонуклеотид-направленный мутагенез
с использованием плазмидной ДНК
Основной недостаток олигонуклеотид-направленного мутагенеза с использованием фага М13 – большое число процедур. Чтобы выделить мутантную форму нужного гена, приходится затратить много времени. В качестве альтернативы системе с использованием фага М13 было разработано множество других подходов, основанных на применении плазмидных ДНК. Это позволяет обойтись без переноса интересующего исследователя гена из плазмиды в фаговую ДНК, а после завершения мутагенеза – обратно в плазмиду. Один из этих подходов включает встраивание ДНК в плазмидный вектор, который несет функциональный ген устойчивости к тетрациклину и неактивный ген устойчивости к ампициллину; в середине последнего заменен один нуклеотид (рис. 8.3). Клетки Е. coli трансформируют вектором, несущим ДНК-мишень, и двухцепочечную плазмидную ДНК денатурируют щелочью с тем, чтобы получить одноцепочечные кольцевые молекулы. Денатурированную ДНК отжигают с тремя разными олигонуклеотидами. Один из них предназначен для внесения изменений в клонированную ДНК-мишень, второй – для устранения мутации в гене устойчивости к ампициллину, третий – для замены одного нуклеотида в гене устойчивости к тетрациклину с тем, чтобы инактивировать этот ген. В реакционную смесь добавляют четыре дезоксирибону клеозидтрифосфата и ДНК-полимеразу Т4, функционирующую аналогично фрагменту Кленова ДНК-полимеразы I Е. coli. Гибридизовавшиеся олигонуклеотиды служат затравками для синтеза ДНК, а интактная кольцевая молекула ДНК – матрицей. Одноцепочечные разрывы в новосинтезированной цепи зашиваются с помощью ДНК-лигазы Т4. По окончании синтеза и лигирования продуктами реакции трансформируют клетки Е. coli. Трансформантов отбирают по признаку устойчивости к ампициллину и чувствительности к тетрациклину. Примерно 90% из них содержат специфическую мутацию в клонированном гене. У остальных трансформантов клонированный ген не был изменен либо потому, что олигонуклеотид не гибридизовался с ним, либо потому, что он вытеснялся в ходе синтеза ДНК. Клетки, несущие мутантный клонированный ген, идентифицируют с помощью гибридизации. Все плазмиды, штаммы, ферменты, олигонуклеотиды (кроме того, который предназначен для изменения клонированного гена), а также буферы продаются в наборе, что облегчает работу.
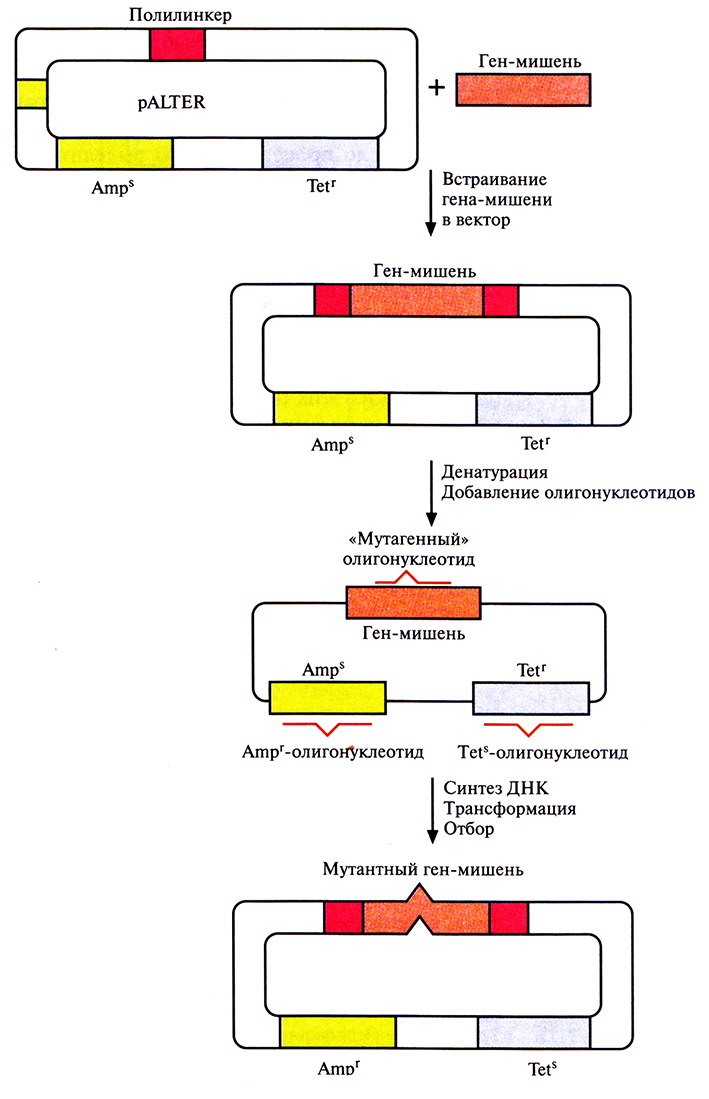
Рис. 8.3. Олигонуклеотид-направленный мутагенез с использованием плазмидной ДНК. Ген-мишень встраивают в полилинкер вектора pALTER. Плазмидную ДНК денатурируют в щелочи и отжигают с тремя олигонуклеотидами «мутагенным» олигонуклеотидом, оли гонуклеотидом, восстанавливающим устойчивость к ампициллину (Аmрr), и олигонуклеотидом, придающим чувствительность к тетрациклину (Tets). Эти олигонуклеотиды служат затравками для синтеза ДНК с помощью ДНК-полимеразы Т4, а исходная цепь – матрицей. Одноцепочечные разрывы в новосинтезированной цепи зашиваются ДНК-лигазой Т4. Продуктами реакции трансформируют клетки Е. coli и отбирают трансформантов Аmрr и Tets.
Олигонуклеотид-направленный мутагенез
с использованием ПЦР-амплификации
Более простой и быстрый метод получения больших количеств мутантных генов, альтернативный системе с использованием фага М13, – сайт-специфический мутагенез в сочетании с полимеразной цепной реакцией (ПЦР). Один из вариантов этого подхода состоит в следующем. Ген-мишень встраивают в плазмидный вектор (рис. 8.4) и помещают препарат в две пробирки. В каждую из них добавляют по два специфических праймера для ПЦР: 1 и 2 в одну пробирку, 3 и 4 – в другую. Праймеры 2 и 3 полностью комплементарны одному из участков клонированного гена или прилегающей к нему последовательности, а 1 и 3 комплементарны другому участку, но содержат один некомплементарный нуклеотид и гибридизуются с разными цепями, так что в результате происходит замена обоих нуклеотидов данной пары. Положение сайтов гибридизации праймеров 1 и 2 в одной пробирке и 3 и 4 – в другой таково, что ПЦР-продукты в разных пробирках имеют разные концы. По окончании ПЦР содержимое пробирок объединяют и проводят денатурацию, а затем ренатурацию. Поскольку концы амплифицированных молекул ДНК из двух пробирок неодинаковы, одноцепочечные ДНК из разных пробирок ассоциируют с образованием кольцевых молекул с двумя одгюцепочечными разрывами. Эти разрывы репарируются in vivo после трансформации Е. coli. При ренатурации одиночных цепей из одной пробирки образуются линейные молекулы. В клетках Е. coli стабильно поддерживаются в виде плазмид и наследуются только кольцевые, а не линейные молекулы, при этом все они несут сайт-специфическую мутацию. Таким образом, с помощью описанного метода можно вносить точковые мутации в клонированный ген, при этом отпадает необходимость во встраивании гена в ДНК фага М13, использовании мутантных штаммов Е. coli типа clut ung и в переносе мутантного гена из М13-вектора в экспрессирующий вектор.
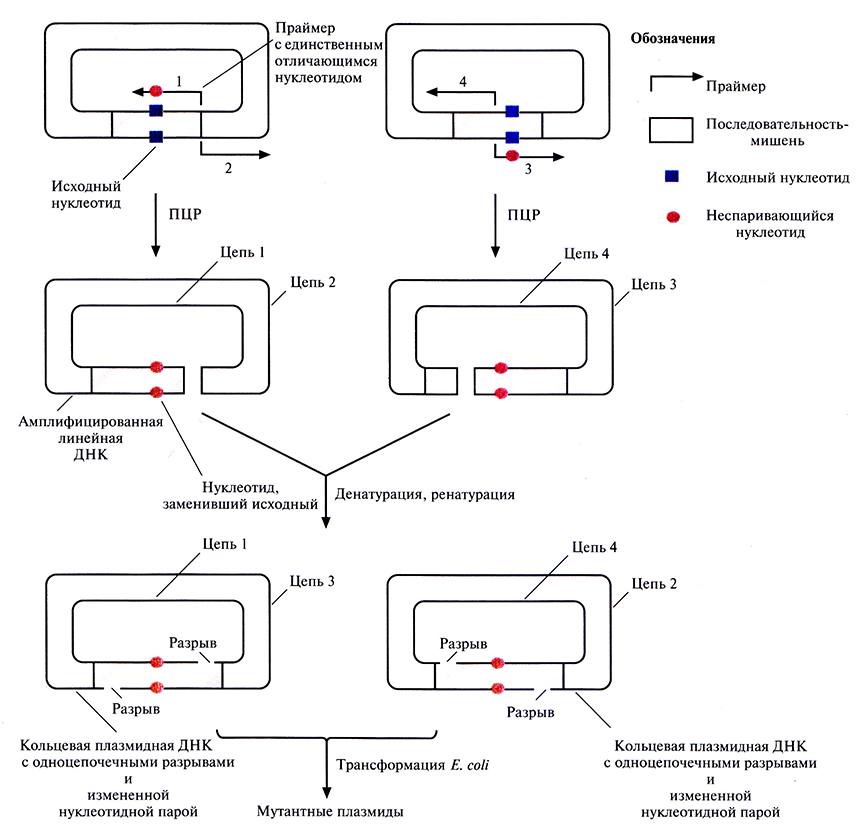
Рис. 8.4. Олигонуклеотид-направленный мутагенез с использованием ПЦР. Реакцию проводят в двух пробирках, в каждой из которых содержится одинаковая двухцепочечная плазмидная ДНК, но разные наборы праймеров. Праймеры 1 и 3 содержат один неспаривающийся нуклеотид и комплементарны разным цепям плазмидной ДНК. Праймеры 2 и 4 полностью комплементарны соответствующим участкам плазмидной ДНК и тоже гибридизуются с разными цепями. Положение сайтов гибридизации для праймеров каждой пары различается, но их концы стыкуются. В результате ПЦР-амплификации образуются линейные молекулы. По окончании реакции содержимое пробирок смешивают и проводят денатурацию, а затем ренатурацию. В результате кроме двух исходных линейных амплифицированных молекул образуются две кольцевые плазмидные ДНК, каждая с двумя одноцепочечными разрывами. После трансформации кольцевыми молекулами клеток Е. coli разрывы репарируются ферментами клетки-хозяина, и плазмида может реплицироваться независимо. Линейные молекулы ДНК в Е. coli не сохраняются.
Случайный мутагенез с использованием
«вырожденных» олигонуклеотидных праймеров
К сожалению, обычно бывает неизвестно, какую нуклеотидную замену в клонированном гене нужно произвести, чтобы получить белок с нужными свойствами. Поэтому часто приходится изменять один определенный нуклеотидный сайт всеми возможными способами. Например, можно синтезировать олигонуклеотидные праймеры, в одном из сайтов которых находятся разные нуклеотиды. Такие «вырожденные» олигонуклеотиды обычно получают, добавляя в автоматический синтезатор ДНК на определенном этапе, когда к цепи должен просоединяться специфический нуклеотид, небольшое количество (до нескольких процентов) трех других нуклеотидов (рис. 8.5). В результате получается гетерогенный по одному сайту набор олигонуклеотидных праймеров, с помощью которых можно получить соответствующий набор мутантных генов-мишеней с нуклеотидными заменами в специфическом сайте.
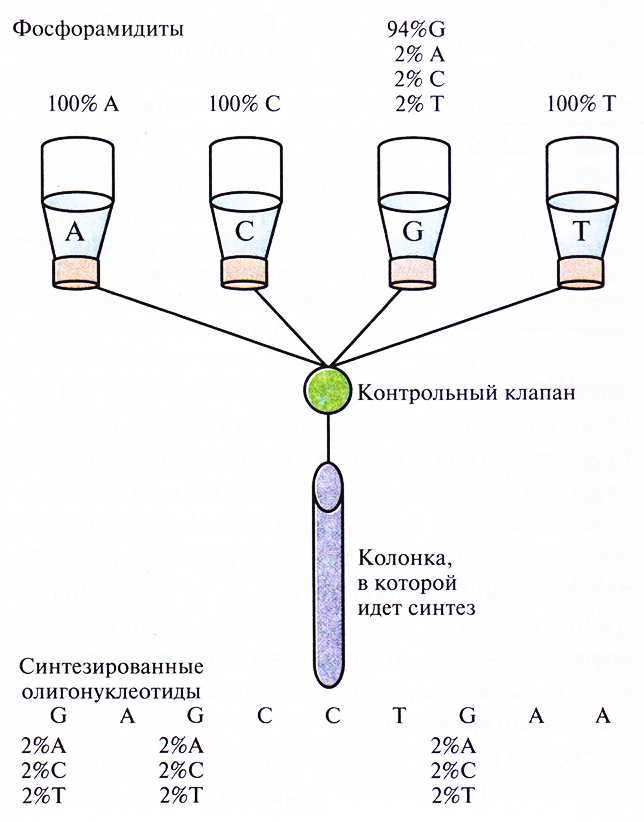
Рис. 8.5. Химический синтез олигонуклеотидных праймеров, содержащих в определенных сайтах разные нуклеотиды. В данном случае в сосуде с G-фосфорамидитом (94%) содержатся также фосфорамидиты А (2%), С (2%) и Т (2%), так что в результате реакции образуется смесь олинонуклеотидов, в которых в тех сайтах, где должен находиться G, присутствуют А, С или Т.
Этот подход имеет два преимущества: 1) не нужно в точности знать, какую роль играет тот или иной аминокислотный остаток в функционировании белка: 2) поскольку в данном сайте происходят разные аминокислотные замены, могут случайно синтезироваться белки с разнообразными интересными и полезными свойствами. Конечно, если ни один из образующихся белков не обладает нужными свойствами, приходится все начинать сначала, синтезировав новый набор «вырожденных» праймеров, комплементарных другой области гена.
ВАЖНАЯ ВЕХА
Олигонуклеотид-направленный мутагенез с использованием векторов на основе фага М13:
эффективный универсальный метод внесения точковых мутаций в любой фрагмент ДНК
М. J. Zoller, М. Smith Nucleic Acids Res. 10: 6487-6500. 1982
Методика олигонуклеотиднаправленного мутагенеза (сайт-специфического мутагенеза) была разработана в основном в лаборатории М. Смита как модификация метода «спасения маркера». При «спасении маркера» мутацию в фаговой ДНК устраняют с помощью отжига мутантной ДНК с фрагментом комплементарной ДНК дикого типа. Было показано, что, отжигая химически синтезированный олигонукпеотид с фаговой ДНК, можно, напротив, вносить в нее мутации. К сожалению, этот и другие методы олигонуклеотид-направленного мутагенеза требовали для своего применения специальных навыков и вначале применялись только в нескольких научно-исследовательских лабораториях. Подход, разработанный Цоллером и Смитом, позволял относительно просто, специфично и быстро вносить мутации в любой клонированный ген и сразу же получил широкое распространение. В его основе лежит использование фага М13 Е. coli. Чужеродную ДНК встраивают в двухцепочечную репликативную форму фаговой ДНК, к одноцепочечной ДНК добавляют олигонуклеотид с необходимыми заменами, получают мутантную копию ДНК, а затем мутантную двухцепочечную форму. Впоследствии эта методика была существенно усовершенствована и упрощена и использовалась для направленного мутагенеза тысяч разных генов.
Частично вырожденные олигонуклеотиды могут быть встроены в ген-мишень разными способами. Один из подходов состоит в следующем. Ген встраивают в плазмиду между двумя уникальными сайтами рестрикции и проводят амплификацию его левого и правого перекрывающихся между собой фрагментов при помощи нескольких ПЦР (рис. 8.6). Пара праймеров, которая используется для амплификации левого фрагмента, включает неполностью комплементарный олигонуклеотид, спаривающийся с тяжелой цепью гена-мишени, и обычный, полностью комплементарный праймер, гибридизующийся с участком легкой цепи, фланкирующим левый уникальный сайт рестрикции. Один из праймеров, использующихся для амплификации правого фрагмента, содержит некомплементарные нуклеотиды и спаривается с тяжелой цепью гена-мишени, а второй праймер полностью комплементарен участку легкой цепи, фланкирующему второй (правый) уникальный сайт рестрикции. Продукты ПЦР-амплификации очищают и объединяют, а затем подвергают денатурации и ренатурации. В результате образуется некоторое количество частично двухцепочечных молекул ДНК, спаренных в области гена-мишени. Их достраивают до полностью двухцепочечных с помощью ДНК-полимеразы, а затем проводят ПЦР-амплификацию с парой праймеров, комплементарных противоположным концам молекул. Амплифицированные молекулы обрабатывают двумя эндонуклеазами рестрикции, уникальные сайты которых находятся на концах фрагмента, и встраивают в соответствующий плазмидный вектор. Этот подход позволяет получить измененные гены со случайными мутациями.
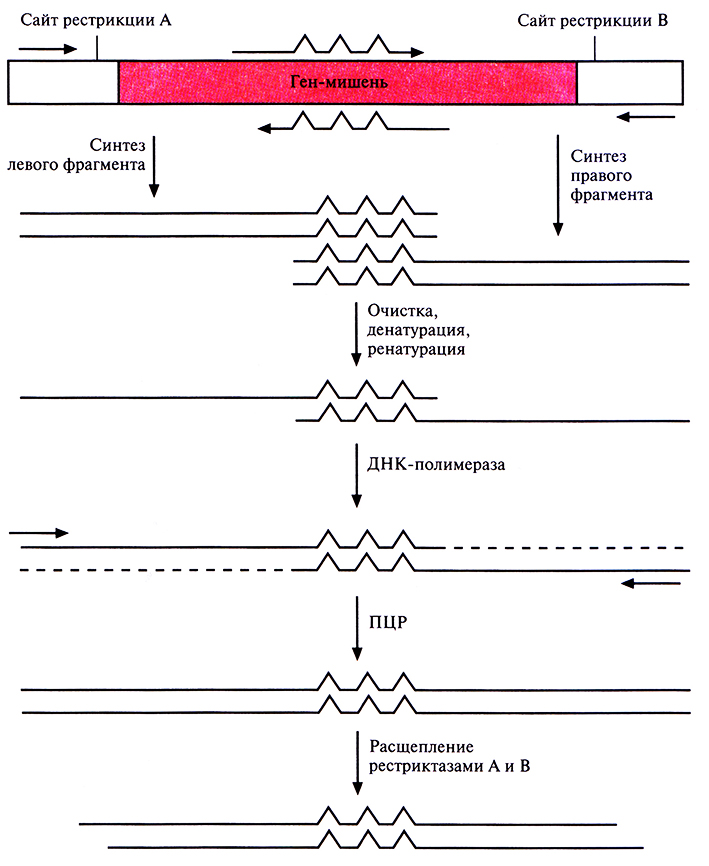
Рис. 8.6. Случайный мутагенез с использованием «вырожденных» олигонуклеотидов и ПЦР. Левую и правую части гена-мишени амплифицируют по отдельности с помощью ПЦР. Соответствующие праймеры показаны горизонтальными стрелками. «Вырожденные» олигонуклеотиды изображены стрелками с тремя зазубринами, каждая из которых отвечает нуклеотиду, не комплементарному соответствующему нуклеотиду в гене-мишени. Амплифицированные фрагменты очищают, денатурируют до полного разделения цепей и ренатурируют. В результате образуются частично двухцепочечные молекулы ДНК, спаренные в области гена-мишени. Их достраивают с помощью ДНК-полимеразы и проводят ПЦР-амплификацию. ПЦР-продукты расщепляют эндонуклеазами рестрикции А и В и встраивают в вектор, обработанный теми же ферментами.
Случайный мутагенез с использованием аналогов нуклеотидов
Помимо методов внесения мутаций в клонированный ген, основанных на использовании фага М13, были разработаны другие подходы, в которых использовались плазмидные ДНК. Один из них схематично представлен на рис. 8.7. Ген-мишень встраивают в плазмиду поблизости от двух тесно расположенных сайтов рестрикции. Эти сайты подбирают так, чтобы после расщепления двумя рестриктазами образовывались укороченные 3'- и 5'-концы, а именно, чтобы 3'-конец сайта расщепления, расположенного рядом с клонированным геном, был укорочен, а 3'-конец с другой стороны плазмиды выступал.
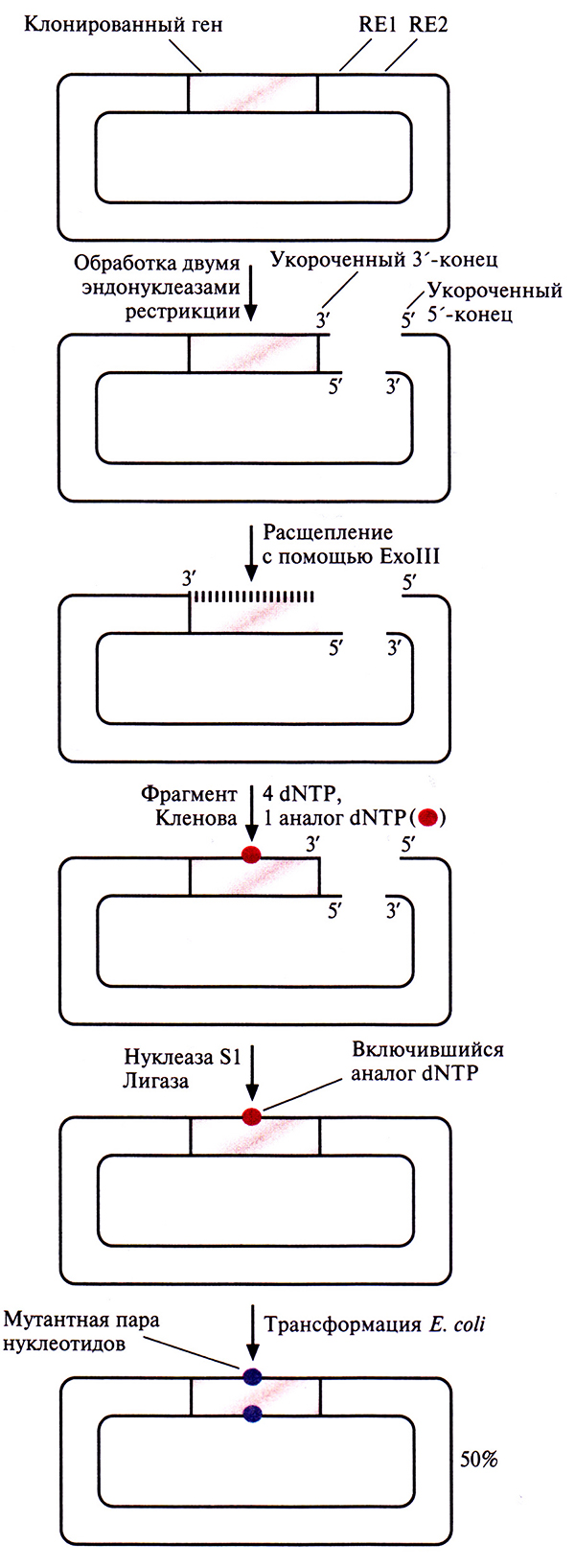
Рис. 8.7. Внесение случайных мутаций в клонированный ген. Вектор, несущий клонированный ген, расщепляют рестриктазами RE1 и RE2, в результате чего образуются один 3'- и один 5'-укороченные концы (и соответственно один 3'- и один 5'-выступающие концы). Затем его обрабатывают ферментом ЕхоIII, который расщепляет ДНК только с укороченного 3'-конца, удаляя по одному нуклеотиду Через некоторое время пеакцию останавливают и заполняют образовавшийся пробел с помощью фрагмента Кленова ДНК-полимеразы I Е. coli. При этом в реакционную смесь добавляют все четыре дезоксинуклеозидтрифосфата (dNTP) и в небольшом количестве – анатогодного из них. Обрабатывают продукт нуклеазой S1 для образования тупых концов, лигируют с помощью ДНК-лигазы Т4 и трансформируют клетки Е. coli. При последующей репликации векторной ДНК в комплементарную цепь включаются нуклеотиды, отличные от исходных, в том сайте, где находится аналог нуклеотида. В результате в клонированный ген вносится мутация.
Экзонуклеаза III (ExoIII) Е. coli расщепляет молекулу ДНК только с укороченных 3 '-концов, но не с выступающих 3'- или любых 5'-концов. Ее добавляют в реакционную смесь после инкубированния ДН К с двумя рестриктазами, и она отщепляет от укороченного 3'-конца цепи по одному нуклеотиду. Через определенное время реакцию останавливают и заполняют пробел с помощью фрагмента Кленова ДНК-полимеразы I, используя смесь обычных четырех дезоксирибонуклеотидов с добавлением аналога одного из них. В результате получают плазмиды, содержащие ген-мишень, в одном или нескольких сайтах которого находится аналог соответствующего нуклеотида. Ими трансформируют клетки Е. coli Плазмиды реплицируются, и в клонированный ген включается нуклеотид, отличный от такового в исходном гене.
Помимо описанного выше, для случайного мутагенеза используют и другие методы, например один из вариантов олигонуклеотиднаправленного мутагенеза с применением ДНК фага М13. В этом случае затравкой для синтеза ДНК служит смесь олигонуклеотидов, содержащих случайные замены. В результате получают библиотеки клонов, несущих множество мутаций в различных сайтах. Недостаток подходов, при которых в клонированном гене образуется большое число случайных мутаций, состоит в необходимости тестирования каждого клона для идентификации того, который детерминировал бы синтез нужного белка. Это весьма непростая задача, но зачастую только так можно выявить белки, обладающие новыми свойствами. Как только эта задача решена, определяют нуклеотидную последовательность соответствующего клонированного гена и идентифицирую! измененный сайт(сайты).
ГЕННАЯ ИНЖЕНЕРИЯ БЕЛКОВ
На долю 20 из многих тысяч изученных и охарактеризованных ферментов приходится более 90% всех ферментов, используемых в настоящее время в промышленности. В табл. 8.1 перечислены некоторые наиболее важные из них и указана область их применения. Остальные ферменты не используются потому, что присущая им активность не удовлетворяет требованиям, предъявляемым высокоспециализированными процессами, протекающими in vitro. Большинство ферментов быстро денатурируют при высокой температуре и в присутствии органических растворителей, а именно в этих условиях протекают многие промышленные процессы. Конечно, термостабильные ферменты можно выделить из термофильных микроорганизмов, однако эти организмы не всегда синтезируют именно те специфические ферменты, которые нужны. Впрочем, эти трудности можно преодолеть при помощи направленного мутагенеза и клонирования генов-мишеней.
Таблица 8.1. Некоторые ферменты и области их применения

Образование дополнительных дисулъфидных связей
Термостабильность белковых молекул можно повысить, внеся в них изменения, благодаря которым они дольше не разворачиваются при повышении температуры. Кроме того, такие термостабильные белки часто не разрушаются в органических растворителях и при нефизиологических условиях (например, при экстремальных рН). К значительному повышению стабильности белковой молекулы может привести образование в ней дополнительных дисульфидных связей. Основная проблема здесь заключается в том, чтобы эти связи не мешали нормальному функционированию белка. В одном из экспериментов при помощи олигонуклеотид-направленного мутагенеза были созданы шесть вариантов лизоцима фага Т4 с новыми внутрицепочечными дисульфидными связями. Для этого два, четыре или шесть специфических аминокислотных остатков в полинуклеотидной цепи были заменены на остатки цистеина, в результате чего образовалась одна, две и три дисульфидных связи соответственно (табл. 8.2).
Таблица 8.2. Свойства лизоцима Т4 и шести его вариантов, полученных с помощью генной инженерии1)
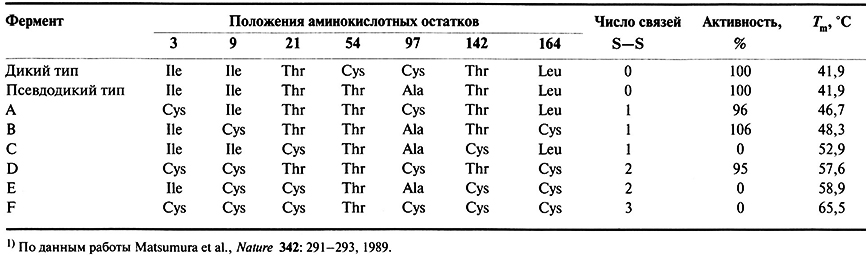
Аминокислотные остатки, замененные на остатки цистеина, располагались в активном ферменте близко друг к другу, так что при образовании новых дисульфидных связей общая конформация молекулы существенно не изменялась. Кроме того, они находились вне активного центра фермента – области, наиболее чувствительной к малейшим изменениям конформации. Связи образовывались между остатками 3 и 97, 9 и 164, 21 и 142 (начало нумерации с N-конца).
После мутагенеза мутантные гены идентифицировали и экспрессировали в Е. coli, рекомбинантные белки очищали и определяли их ферментативную активность и термостабильность (табл. 8.2). Последняя обычно характеризуется температурой, при которой молекула денатурирует на 50%; степень денатурации определяется по изменению кругового дихроизма раствора белка. Исходная (нативная) форма лизоцима Т4 содержит два свободных остатка цистеина, не участвующих в образовании дисульфидных связей. У фермента так называемого «псевдодикого типа» они заменены на остатки Thr и Ala, при этом активность и термостабильность фермента остались прежними. Последовательность «псевдодикого типа» служила стандартом при сравнении вариантов с потенциально термостабилизирующими дисульфидными связями и также предотвращала образование случайных дисульфидных связей между вновь введенными остатками цистеина и остатками, присутствующими в нативном белке.
Результаты этого эксперимента показали, что термостабильность фермента повышается при образовании новых дисульфидных связей, при этом наиболее термостабильным является белок с максимальным числом таких связей. Однако некоторые варианты (С, Е и F), будучи более термостабильными, чем нативный фермент или фермент «псевдодикого типа», не обладают ферментативной активностью. Возможно, это обусловливается искажением конформации белковой молекулы при образовании дисульфидной связи между остатками 21 и 142. Хотя создание новых белков с помощью методов генной инженерии часто представляет собой эмпирический процесс (т. е. далеко не всегда бывает ясно, замены каких именно аминокислот позволяют получить «наилучший» вариант), описанный эксперимент показывает, что получение термостабильных белков с дополнительными дисульфидными связями вполне реально.
Была предпринята также попытка получения термостабильной мутантной ксиланазы Bacillus circulans – фермента, который можно использовать при производстве бумаги. Одним из этапов этого процесса является удаление гемицеллюлозы из пульпы с целью ее отбеливания, при этом образуются большие количества токсичных отходов. Обработка древесной массы ксиланазой позволяет использовать меньше отбеливающих химикатов. К сожалению, перед добавлением фермента пульпу обрабатывают горячей щелочью, а поскольку современные технологии имеют тенденцию к уменьшению количества воды, расходуемой на охлаждение пульпы, ксиланаза должна оставаться активной при относительно высоких температурах. Чтобы определить, в какие участки полипептидной цепи могут быть введены одна, две или три дисульфидных связи для стабилизации фермента без нарушения его каталитической активности, использовали компьютерное моделирование пространственной структуры ксиланазы. Были получены восемь производных ксиланазы В. circulans. Все они обладали более высокой термостабильностью, чем нативный фермент, и при этом три были столь же активны при 60 °С, как и нативный белок, а один, содержащий дисульфидную связь между N- и С-концами, был даже в два раза более активным и сохранял свыше 85% своей активности после 2-часовой инкубации при 60 °С, в то время как нативный фермент полностью утрачивал активность в этих условиях уже через 30 мин. Успех этих экспериментов показывает, что данную стратегию можно использовать для повышения термостабильности различных ферментов, если только для них имеются достаточно полные рентгеноструктурные данные. И тем не менее пока нельзя быть уверенным, что термостабильная ксиланаза будет широко использоваться при производстве бумаги.
Замена аспарагина на другие аминокислоты
При высоких температурах остатки аспарагина и глутамина могут дезамидироваться с образованием аммиака. Теряя амидную группу, они превращаются в аспарагиновую и глутаминовую кислоты соответственно, что приводит к локальным изменениям конформации полипептидной цепи и как следствие – к утрате активности белков, в которые они входят.
Чтобы установить, какое влияние оказывает замена некоторых остатков аспарагина в молекуле триозофосфат-изомеразы Saccharomyces cerevisiae на свойства фермента, были поставлены специальные эксперименты. Триозофосфат-изомераза состоит из двух идентичных субъединиц; каждая из них содержит два остатка аспарагина, замена которых может приводить к изменению термочувствительности белка, поскольку они расположены в месте соприкосновения субъединиц. При помоши олигонуклеотид-направленного мутагенеза были заменены остатки аспарагина в положениях 14 и 78 (табл. 8.3). Замена одного из них на остаток треонина или изолейцина приводила к повышению термостабильности фермента, на аспарагиновую кислоту – к понижению. Фермент, получающийся при замене обоих остатков аспарагина на остатки аспарагиновой кислоты, оказался нестабильным даже при нормальной температуре и обладал низкой ферментативной активностью (в табл. 8.3 не представлен).
Таблица 8.3. Стабильность при 100 °С триозофосфат-изомеразы дрожжей
и ее генноинженерных вариантов1)

Оценка чувствительности рекомбинантных белков к протеолитическому расщеплению показала, что существует положительная корреляция между термостабильностью белка и его устойчивостью к" 11ротеолитическому расщеплению. Полученные данные говорят о возможности создания термостабильных форм других ферментов путем замены несущественных остатков аспарагина.
Уменьшение числа свободных сулъфгидрилъных групп
Чужеродный белок, синтезируемый в организме-хозяине, иногда оказывается менее активным, чем ожидалось, и чтобы повысить его активность, можно использовать методы генной инженерии. Например, при экспрессии в Е. coli клонированной комплементарной ДНК (кДНК) β-интерферона человека (ИФβ) белковый продукт обладал в 10 раз меньшей противовирусной активностью, чем нативная гликозилированная форма. При этом ИФβ синтезировался в довольно большом количестве, однако почти все его молекулы образовывали димеры и более высокомолекулярные неактивные комплексы.
Как показал анализ нуклеотидной последовательности гена ИФβ, в нем присутствуют три остатка цистеина, и один из них или несколько, возможно, участвуют в образовании дисульфидных связей, приводящих к образованию димеров и олигомеров в клетках Е. coli, но не в клетках человека. Было высказано предположение, что замена одного или нескольких цистеиновых кодонов на сериновые приведет к синтезу интерферона, не образующего олигомеров. Серин был выбран потому, что его структура сходна со структурой цистеина за исключением того, что вместо серы он содержит кислород и поэтому не может образовывать дисульфидные связи.
Приступая к этим экспериментам, исследователи не располагали детальней информацией относительно молекулярной структуры β-интерферона и были вынуждены опираться на соответствующие данные для родственных белков. Иными словами, они не знали, какой из трех остатков цистеина был ответствен за формирование межмолекулярных дисульфидных связей. К счастью, локализация остатков цистеина, участвующих в образовании внутримолекулярных дисульфидных связей в молекуле ИФα с аналогичной структурой, была известна, что делало возможным сравнение аминокислотных последовательностей этих двух молекул (рис. 8.8). Как показали результаты анализа, остатки Cys-31 и Cys-141 в ИФР находятся в тех же позициях что и остатки Cys-29 и Cys-138 в ИФα. Поскольку последние участвуют в образовании в ИФα: внутримолекулярных дисульфидных связей, было разумно предположить, что Cys-17 в ИФβ не вовлечен в формирование таких связей и его можно заменить.
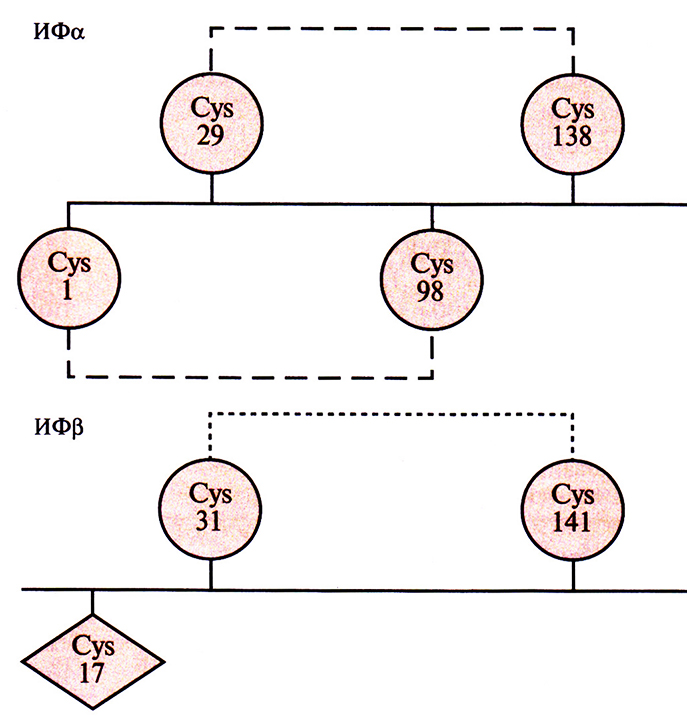
Рис. 8.8. Локализация остатков цистеина в молекулах ИФα и ИΦβ, между которыми образуются дисульфидные связи. Выявленные внутримолекулярные дисульфидные связи в ИФα обозначены штриховыми линиями, а предполагаемая связь в ИФβ — пунктирной.
Это предположение оказалось правильным: при синтезе в клетках Е. coli Ser-17-ИФβ мультимерные комплексы не образовывались. Кроме того, этот интерферон обладал такой же удельной активностью, как и аутентичный нативный ИФβ, и был более стабилен при длительном хранении, чем нативная форма.
Повышение ферментативной активности
С помощью направленного мутагенеза можно не только повышать стабильность ферментов, но и изменять их каталитическую активность. В настоящее время для существенного изменения ферментативной активности любого достаточно хорошо охарактеризованного фермента необходимо располагать детальной информацией о геометрии его активного центра. В этом случае можно предсказать, какие замены необходимо произвести для изменения специфичности фермента к данному субстрату.
Возможности данного подхода иллюстрируют результаты эксперимента по изменению специфичности связывания субстрата тирозил-тРНК–синтетазой из В. stearothermophilus. Этот фермент катализирует аминоацилирование тРНК, которая специфически связывает тирозин (тРНКТуr), в ходе двухступенчатой реакции:
(1) Туr + АТР→Туr-А + РРi
(2) Туr-А + тРНКТуr → Туг-тРНКТуr +АМР
На стадии 1 АТР активирует тирозин (Туr), в результате чего образуется связанный с ферментом тирозиладенилат (Tyr-А) и пирофосфат (PPj). На стадии 2 тирозиладенилат гидролизуется при участии свободной З'-гидроксильной группы молекулы тРНК, так что тирозин присоединяется к тРНК с высвобождением AMP. В ходе обеих реакций субстраты остаются связанными с тирозил-тРНК–синтетазой.
Ко времени постановки эксперимента была определена пространственная структура тирозил-тРНК–синтетазы В. stearothermophilus и локализован ее активный центр, так что при помощи компьютерного моделирования можно было предсказать влияние замены в нем одного или нескольких аминокислотных остатков на взаимодействие фермента с субстратами. Чтобы проверить правильность прогнозов, с помощью олигонуклеотид-направленного мутагенеза в ген тирозил-тРНК–синтетазы были внесены специфические мутации. Остаток треонина в положении 51 (Thr-51) был заменен на остаток аланина или пролина. В нативном ферменте гидроксильная группа Thr-51 образует водородную связь с атомом кислорода рибозного кольца тирозиладенилата. и предполагалось, что разрыв этой слабой связи увеличит сродство фермента к АТР.
Чтобы охарактеризовать получившиеся ферменты, определили их кинетические константы. В некоторых случаях изменения оказались более существенными, чем ожидалось (табл. 8.4). Так, если для А1а-51-фермента константа связывания (Км) с АТР уменьшилась примерно в два раза без значительного изменения каталитической константы (kсаr), то для Рго-51 -фермента – более чем в 100 раз. При этом каталитическая эффективность (kcat/KM) реакции аминоацилирования увеличилась в обоих случаях. Результат, полученный для Рго-51-фермента, был неожиданным, поскольку замена треонина на пролин должна была привести к нарушению (по крайней мере локальному) структуры а-спирали в этой области, что предположительно должно отрицательно сказаться на связывании субстрата.
Таблица 8.4. Эффективность аминоацилирования, осуществляемого нативной (Thr-51)
и модифицированной (Аlа-51 и Рго-51) тирозил-тРНК–синтетазами1'

Эти данные показывают, что несмотря на всю сложность прогнозирования результата специфических аминокислотных замен, с помощью описанного подхода все же можно идентифицировать боковые группы, замена которых приведет к улучшению кинетических свойств фермента. Кроме того, стало очевидно, что сродство данного фермента к субстрату, а также каталитическую эффективность реакции можно повысить in vitro, внося соответствующие изменения в клонированный ген.
Изменение потребности ферментов
в металлических кофакторах
Субтилизины – сериновые протеиназы, секретируемые в культуральную среду грамположительными бактериями, – широко используются в качестве биодеградируемых детергентов. Все они прочно связывают один или несколько атомов кальция, повышающих их стабильность. К сожалению, субтилизины используются в таких промышленных процессах, где участвуют в больших количествах соединения, хелатирующие металлы, в том числе кальций, и в таких условиях субтилизины быстро теряют свою активность. Чтобы решить эту проблему, попытались сначала лишить субтилизин способности связывать кальций, а затем увеличить стабильность модифицированного фермента.
Получение модифицированного субтилизина было начато с идентификации гена BPN Bacillus amyloliquefaciens. Прежде всего с помщью рентгеноструктурного анализа высокого разрешения была определена структура белка, а затем с использованием олигонуклеотид-направленного мутагенеза создан мутантный ген с делетированными нуклеотидами, кодирующими участок белковой молекулы от 75 до 83 аминокислотного остатка, который отвечал за связывание кальция. Белок с делецией не связывал кальций и, что удивительно, сохранял конформацию, сходную с таковой исходного белка.
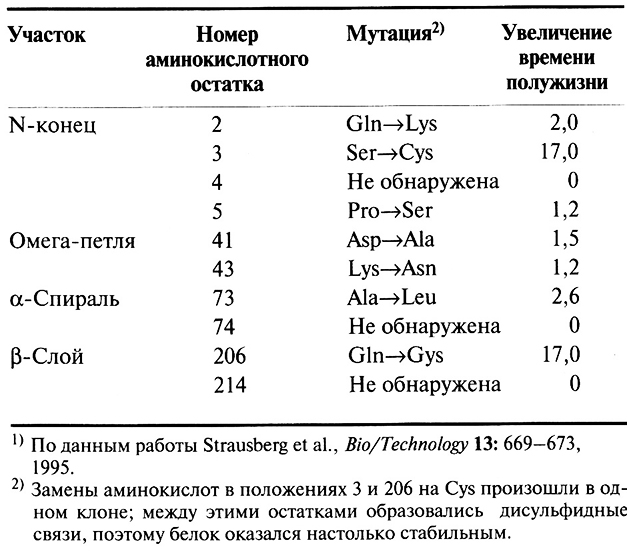
Далее для повышения стабильности субтилизина без кальцийсвязывающего домена идентифицировали сайты, которые необходимо изменить, и аминокислотные остатки, которые нужно ввести в эти сайты. Было высказано предположение, что все аминокислоты, взаимодействующие с кальцийсвязывающим доменом в исходном ферменте, не являются оптимальными в новых условиях. В качестве кандидатов были выбраны 10 аминокислот, а поскольку заранее не было известно, включение какого именно аминокислотного остатка приведет к наибольшей стабилизации фермента, для внесения изменений в каждый из 10 сайтов использовали случайный мутагенез.
Выбранные аминокислоты были локализованы в четырех разных областях белковой молекулы: N-концевом участке (остатки со 2 по 5), в омега-петле (остатки с 36 по 44), в α-спиральой области (остатки с 63 по 85) и в одном из β-слоев (остатки с 202 по 220). Чтобы идентифицировать оптимальную для данного сайта аминокислоту, мутантные клоны выращивали в плашках, прогревали при 65 °С в течение 1 ч, охлаждали и измеряли активность субтилизина. Для синтеза активного, не связывающего кальций субтилизина пришлось использовать В. subtilis, поскольку для Е. coli такой белок оказался токсичным.
Предварительный анализ выявил наличие стабилизирующих мутаций в 7 из 10 исследованных сайтов (табл. 8.5). Фермент, полученный при их внесении в один ген, обладал кинетическими свойствами, сходными с таковыми исходного субтилизина. Кроме того, мутантный субтилизин в отсутствие кальция был почти в 10 раз более стабилен, чем исходный и, как это ни удивительно, примерно на 50% более стабилен, чем исходный фермент в присутствии кальция. Полученные результаты показывают, что несмотря на трудоемкость этих экспериментов с помощью генной инженерии можно изменять свойства ферментов, которые зависят от большого числа аминокислотных остатков.
Таблица 8.5. Влияние случайных замен аминолслотных остатков в определенных участках
на стабильность субтилизина BPN', лишенного кальцийсвязывающего домена1)
Изменение специфичности фермента
Олигонуклеотид-направленный мутагенез используют в основном для улучшения уже существующих свойств ферментов, но, вероятно, с его помощью можно изменять ферменты таким образом, чтобы они преобретали другую специфичность. Например, таким способом на основе относительно неспецифичной эндонуклеазы Fokl были получены новые сайтспенифичные эндонуклеазы.
К настоящему времени идентифицировано более 2500 ферментов рестрикции-модификации, происходящих из большого числа разных организмов. Многие из них узнают одну и ту же нуклеотидную последовательность, так что всего существует около 200 разных рестриктазных сайтов, при этом размер большинства из них составляет от 4 до 6 п. н. Эндонуклеазы рестрикции, узнающие такие сайты, расщепляют молекулу ДНК в очень многих местах и используются для получения больших фрагментов ДНК не столь широко, как эндонуклеазы рестрикции, узнающие нуклеотидные последовательности длинной в 8 п. н. или больше. Поиск новых эндонуклеаз рестрикции – весьма непростая задача, для ее решения требуется много времени. Вряд ли можно надеяться, что удастся найти достаточно много ферментов, узнающих сайты длинной 8 п. н. и больше, так что для получения новых рестриктаз необходимо использовать альтернативные генноинженерные подходы.
Существует весьма интересный класс белков, в молекуле которых присутствуют уникальные структурные домены, связывающие атомы Zn2+, – так называемые цинковые пальцы. Эти белки связываются со специфической нуклеотидной последовательностью, встраиваясь своим α-спиральным участком в большую бороздку двойной спирали. Так, белок Zif268 из клеток мышей содержит три цинковых пальца, каждый из которых взаимодействует с определенным кодоном ДНК. Поскольку эти пальцы связываются с ДНК независимо друг от друга, их можно объединить в составе одного пептида таким образом, чтобы связывание происходило с определенным сайтом. Это позволяет создавать нуклеазы, расщепляющие ДНК в уникальных сайтах, объединив нуклеотидные последовательности, кодирующие цинковые пальцы, с частью гена неспецифичной нуклеазы Fokl бактерии Flavobacterium okeanokoites. Чтобы проверить реальность этого предположения, был создан химерный ген, кодирующий участок из шести остатков гистидина на N-конце белковой молекулы для упрощения очистки рекомбинантного белка, три цинковых пальца, линкер (Gly4Ser)3 для придания гибкости рекомбинантной молекуле, а также содержащий часть гена нуклеазы Fokl (рис. 8.9). После очистки рекомбинантного белка N-концевые остатки гистидина были удалены обработкой тромбином.

Рис. 8.9. Генноинженерная конструкция, кодирующая рекомбининтный белок «цинковые пальцы – эндонуклеаза рестрикции Fokl».
Бактерии, синтезирующие эндонуклеазы рестрикции, защищают собственную ДНК от расщепления с помощью ферментов, метилирующих те участки молекулы, с которыми связывается соответствующая эндонуклеаза рестрикции. Однако геном клетки-хозяина не защищен от рекомбинантной рестриктазы Fokl, и чтобы предотвратить гибель растущих клеток, синтез гибридного фермента подавляли, поместив ее ген под контроль системы экспрессии бактериофага Т7.
В результате этих экспериментов были получены две рекомбинантные эндонуклеазы рестрикции Fokl. Одна из них расщепляла ДНК фага X в том сайте, который и ожидался, а вторая – в ожидаемом сайте и – в меньшей степени – в двух других сайтах. Это не удивительно, поскольку цинковые пальцы распознают в основном два из трех оснований триплета. Хотя эти рекомбинантные ферменты пока нельзя использовать в лаборатории, описанный подход создания уникальных эндонуклеаз рестрикции представляется весьма перспективным.
Таблица 8.6. Стабильность и активность различных мутантных вариантов фермента tPA1), 2)

Повышение стабильности и специфичности фермента
Фермент, называемый активатором тканевого плазминогена (tPA), – это сериновая протеиназа, состоящая из нескольких доменов; ее используют в клинике для растворения сгустков крови. К сожалению, tPA быстро выводится из системы кровообращения, поэтому его приходится вводить путем инфузии. Чтобы добиться желаемого терапевтического эффекта, необходимо использовать высокие концентрации фермента, а это может приводить к неспецифическому внутреннему кровотечению. Таким образом, было бы весьма желательно получить долгоживущий фермент tPA, обладающий высоким сродством к фибрину в тромбах и не вызывающий кровотечения. Белок с такими свойствами можно получить, внося специфические мутации в ген нативного tPA. Заменив Thr-103 на Asn, получили фермент, сохраняющийся в плазме кролика примерно в 10 раз дольше, чем нативный вариант. Заменив аминокислоты 296–299 с Lys-His-Arg-Arg на Аlа-Аlа-Аlа-Аlа. добились существенного повышения сродства фермента к фибрину. Заменив Asn-117 на Gin, получили фермент с такой же фибринолитической активностью, как у исходного фермента. Внеся эти три мутации в один белок, получили фермент, обладающий всеми тремя свойствами (табл. 8.6). Чтобы выяснить, можно ли использовать его вместо нативного tPA, нужно провести дополнительные исследования.
ЗАКЛЮЧЕНИЕ
Свойства любого белка зависят от его конформации которая в свою очередь определяется аминокислотной последовательностью. Некоторые аминокислоты в полипептидной цепи играют ключевую роль в определении специфичности, термостабильности и других свойств белка, так что замена единственного нуклеотида в гене, кодирующем белок, может привести к включению в него аминокислоты, приводящему к понижению его активности, либо, напротив, к улучшению каких-то его специфических свойств. С развитием технологии рекомбинантных ДНК появилась возможность производить специфические замены в клонированных генах и получать белки, содержащие нужные аминокислоты в заданных сайтах. Такой подход получил название направленного мутагенеза. Как правило, интересующий исследователя ген клонируют в ДНК фага М13. Одноцепочечную форму ДНК этого фага копируют с использованием олигонуклеотидного праймера, синтезированного таким образом, чтобы в ген-мишень был встроен определенный нуклеотид Затем трансформируют двухцепочечными ДНК М13 клетки Е. coli. Часть образующихся в клетках фаговых частиц несет ген, содержащий нужную мутацию. Такие частицы идентифицируют, встраивают мутантный ген в экспрессирующий вектор, синтезируют белок и определяют его активность. Вносить изменения в клонированные гены можно также с помощью плазмид или ПЦР. Обычно заранее не известно, какую именно аминокислоту (аминокислоты) необходимо заменить для того, чтобы улучшить то или иное свойство белка-мишени. Поэтому предпочтительно использовать случайный, а не олигонуклеотид-направленный мутагенез.
Выбор аминокислоты, подлежащей замене, как правило, производится с учетом ее роли в функционировании белка. Данные об этом получают в ходе генетических исследований или методом рентгеноструктурного анализа трехмерной структуры белка. Изменяя специфические сайты или целые участки белковой молекулы, можно повысить термостабильность белка, изменить его чувствительность к рН, специфичность, аллостерическую регуляцию, потребность в кофакторе и другие свойства. Так, термостабильность триозофосфатиозомеразы удалось повысить, заменив аминокислоты в двух позициях. Этот подход можно использовать как для придания новых свойств уже существующим белкам, так и для создания уникальных ферментов.
