Долгое время медицинская генетика занималась одной проблемой: установлением генетических основ наследственных заболеваний человека. Были разработаны диагностические тесты для выявления ряда таких заболеваний у новорожденных или плода, и по их результатам проводилось генетическое консультирование семей, относящихся к группе риска. Это позволяло подготовить консультируемых к возможности проявления данного заболевания у их потомков. Кроме того, иногда удавалось смягчить негативные последствия генетического дефекта с помощью медикаментозной терапии, переливания крови или назначения диеты.
Нормальная работа организма обеспечивается функциями множества взаимосвязанных генов, и мутация даже в одном из них может иметь самые разные последствия. Так, мутация, в результате которой изменяется активность того или иного фермента, может приводить к накоплению токсичного субстрата или дефициту соединения, необходимого дта нормального функционирования клетки, а мутация в гене, кодирующем структурный белок, – к серьезным нарушениям на уровне клеток, тканей или органов. Кроме того, мутация в гене, экспрессирующемся в одной ткани, может сказаться самым серьезным образом на совсем другой ткани и привести к появлению множества симптомов. Например, мутация в гене печеночного фермента фенилалалиндегидроксилазы, в результате которой блокируется превращение фенилаланина в тирозин, оказывает серьезное влияние на нервную систему. У индивидуума, гомозиготного по дефектному гену, этот фермент не вырабатывается вообще или вырабатывается в очень небольших количествах; это приводит к повышению уровня эндогенного фенилаланина в крови, к неправильному формированию миелиновой оболочки вокруг аксонов нервных клеток центральной нервной системы и как следствие – к тяжелой умственной отсталости. Это врожденное заболевание, называемое фенилкетонурией, встречается у европеоидов с частотой 1 на 10 000 новорожденных. В каждой ткани организма экспрессируется свой набор из всей совокупности генов, но есть мутации, которые приводят к болезням, затрагивающим буквально все органы и ткани: мышцы, глаза, печень, кости, почки, сердце, нервную систему, кожу, мозг, желудок, кишечник, систему кроветворения.
Конечной целью медико-генетических исследований является создание методов лечения всех наследственных заболеваний. В табл. 21.1 перечислены продукты некоторых генов, коротко описаны симптомы заболеваний, которые обусловливаются мутациями в этих генах, указаны способы их лечения. Наследственные заболевания имеют сложные клинические проявления, и их лечение носит во многом симптоматический характер. Некоторые нарушения метаболизма корректируют назначением специальной диеты, что приводит к снижению уровня токсичных веществ в организме, накопление которых обусловливается мутациями в определенных генах. Так, при фенилкетонурии, которую выявляют у новорожденных с помощью специфического биохимического анализа крови, назначают безаланиновую диету. Для облегчения симптомов наследственных заболеваний, связанных с дефектом определенного белка, вводят внутривенно его функциональную форму, не вызывающую иммунной реакции. Такую «заместительную» терапию используют, например, для лечения гемофилии, тяжелого комбинированного иммунодефицита (SCID, от англ. severe combined immunodeficiency) и болезни Гоше. Иногда для компенсации каких-то утраченных функций проводят трансплантацию костного мозга или других органов. К сожалению, нередко интенсивное лечение многих наследственных болезнЛ начинают проводить только тогда, когда пациент находится в критическом состоянии и удается лишь ненамного продлить его жизнь. Поскольку генетические заболевания часто носят системный характер и постепенно приводят к ослаблению организма, разработка эффективных методов лечения представляет собой непростую задачу. Существующая терапия, как правило, малоэффективна, лишь немногие пациенты доживают до старости и могут иметь детей. В большинстве случаев лечение необходимо проводить многократно, оно очень дорогое и длительное. Поэтому разработка новых видов терапии очень актуальна.
Таблица 21.1. Применяемые в настоящее время методы лечения некоторых моногенных заболеваний человека
После того как были установлены молекулярные основы трансформации бактерий (переноса генов из одного штамма в другой), у ученых появилась надежда, что аналогичный механизм – введение нормальных генов в дефектные соматические клетки – можно будет использовать для лечения наследственных заболеваний человека. Перспективы генной коррекции соматических клеток стали более реальными в 1980-х гг.; к этому времени были разработаны методы получения изолированных генов, созданы эукариотические экспрессирующие векторы, стали рутинными эксперименты по переносу генов на мышах. В 1990 г. была предпринята первая попытка применения генотерапии для лечения SCID (табл. 21.1) у двух девочек.
Использовался следующий подход. Клонированную кДНК аденозиндезаминазы (АДА) ввели в лимфоциты, полученные от каждой из пациенток. Модифицированные клетки, синтезирующие АДА, культивировали и в течение двух лет с определенной периодичностью вводили девочкам. Через четыре года после начала лечения у обеих пациенток наблюдалась экспрессия гена АДА и отмечалось облегчение симптомов SCID. Однако истинная причина улучшения осталась не совсем ясной: был ли это эффект заместительной терапии (внутривенного введения защищенной формы АДА – полиэтиленгликоль-АДА) или собственно генной терапии. Беспорно одно: этот опыт показал безопасность генной терапии. Сходные результаты были получены и для других пациентов с SCID, одновременно получавших оба вида лечения. Исследования были продолжены на большем числе больных.
В соответствии с законодательством США, прежде чем новый лекарственный препарат будет разрешен к применению, он должен пройти четыре строго оговоренных стадии проверки.
1. Доклинические испытания, которые включают многочисленные эксперименты, проводимые in vitro и на лабораторных животных.
2. I фаза клинических испытаний проводится на небольшом числе (от 6 до 10) пациентов и часто имеет целью проверку безопасности препарата.
3. II фаза клинических испытаний проводится на большем числе пациентов и имеет целью проверку эффективности действия препарата.
4. Ill фаза клинических испытаний проводится с привлечением большого числа испытуемых и включает исчерпывающий анализ надежности и эффективности препарата, при этом используется информация, полученная на предыдущих этапах.
Прежде чем начать проверку препарата, необходимо, чтобы протокол его испытаний был одобрен и утвержден в соответствующих контролирующих инстанциях. С 1990 по 1992 г. было одобрено более десяти протоколов испытаний по генной терапии, находящихся в I фазе, а к 1997 г. – более 200 протоколов испытаний по генной терапии разных видов злокачественных новообразований, гемофилии, СПИДа, муковисцидоза, гиперхолестеролемии, бокового амиотрофического склероза и др. (табл. 21.2). Прежде чем приступать к I фазе клинических испытаний, необходимо учесть ряд важных моментов: предполагаемое исследование должно быть направлено на разработку методов лечения однозначно диагностируемой болезни, соответствовать существующим правилам проведения медико-биологических экспериментов и осуществляться с минимальным для пациента риском.
Таблица 21.2. Некоторые заболевания, генная терапия которых проходит испытания с 1990 г.

Поскольку генная терапия представляет собой новое направление, а заболевания, которые предполагают лечить с ее помощью, столь различны, рассматривают множество разных подходов. В настоящее время все исследования по генной терапии направлены на коррекцию генетических дефектов соматических, а не половых (зародышевых) клеток. Это объясняется этическими и чисто техническими причинами, а также соображениями безопасности: ведь ДНК, введенная в половые клетки человека, передавалась бы последующим поколениям.
В самом общем смысле под генной терапией соматических клеток человека понимают коррекцию специфического наследственного заболевания путем введения в клетку-мишень функционального экспрессирующегося гена. Однако за этим простым определением скрывается целый ряд проблем. Например, как получить доступ к клеткам, предназначенным для коррекции? Как осуществить доставку терапевтического гена? Какая доля клеток-мишеней должна получить такой ген, чтобы болезнь отступила? Необходим ли точный контроль транскрипции введенного гена для обеспечения ее эффективности? Не вызовет ли избыточная экспрессия введенного гена побочных эффектов? Будут ли модифицированные клетки поддерживаться бесконечно или потребуются повторные введения?
Хотя генная терапия соматических клеток делает только свои первые шаги, на ряд вопросов, касающихся некоторых наследственных заболеваний, уже получены ответы. Появляются все новые подходы к генной терапии соматических клеток, которые можно разделить на две большие категории: генная терапия in vivo и ex vivo. Разрабатывают и специфические лекарственные препараты на основе нуклеиновых кислот: антисмысловые олигонуклеотиды; РНК-ферменты, модифицированные с помощью генной инженерии; олигонуклеотиды, корректирующие генные мутации in vivo.
ГЕННАЯ ТЕРАПИЯ EX VIVO
Генная терапия ex vivo, как правило, включает следующие этапы (рис. 21.1).
1. Получение клеток от больного.
2. Исправление генетического дефекта с помощью переноса нужного гена в изолированные клетки.
3. Отбор и наращивание генетически «исправленных» клеток.
4. Инфузия или трансплантация этих клеток пациенту.
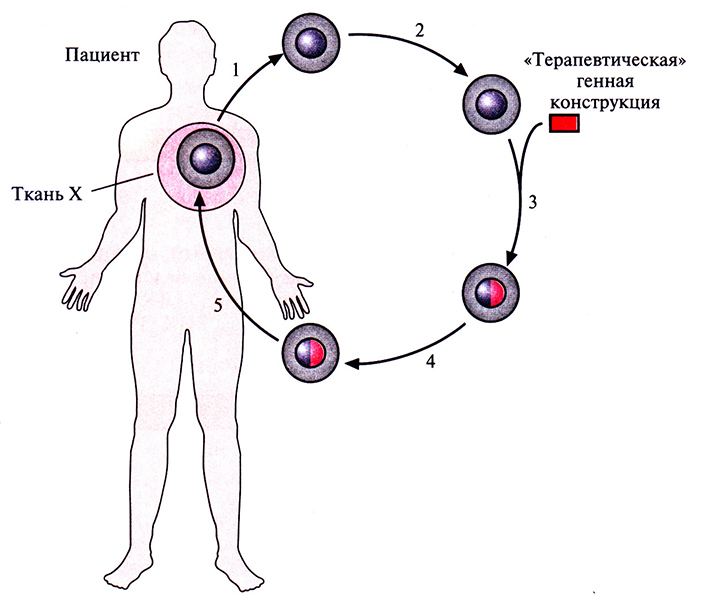
Рис. 21.1. Схематическое представление генной терапии ex vivo. Процедура включает: 1) получение от пациента клеток с генным дефектом; 2) культивирование изолированных клеток; 3) трансфекцию «терапевтической» генной конструкции в изолированные клетки; 4) отбор выращивание и тестирование трансфицированных клеток; 5) трансплантацию или трансфузию трансфицированных клеток пациенту.
Использование собственных клеток пациента (аутологичных клеток) гарантирует, что после инфузии или трансплантации у него не разовьется иммунный ответ.
Необходимо, чтобы процедура переноса генов, используемая для генной терапии ex vivo,была эффективной, а «терапевтический» ген стабильно поддерживался и непрерывно экспрессировался. Этим условиям отвечают векторы, полученные на основе мышиных ретровирусов. Но ретровирусы имеют существенный недостаток – они могут приводить к злокачественной транформации клеток. Такую вероятность необходимо уменьшить, а лучше полностью исключить.
Геном ретровируса дикого типа представлен двумя идентичными одноцепочечными молекулами РНК, каждая из которых состоит из шести участков: длинного концевого повтора (5'-LTR, от англ. long terminal repeat); некодируюшей последовательности пси+ (ψ+), необходимой для упаковки РНК в вирусную частицу; трех генов, кодирующих структурный белок внутреннего капсида (gag), белок, обладающий функциями обратной транскриптазы и интегразы (pol), и белок оболочки (env); 3'-LTR-последовательности (рис. 21.2). Жизненный цикл ретровируса включает следующие стадии.
1. Инфицирование клетки-мишени.
2. Синтез ДНК-копии генома с помощью собственной обратной транскриптазы.
3. Транспорт вирусной ДНК в ядро.
4. Встраивание вирусной ДНК в один из хромосомных сайтов клетки-хозяина.
5. Транскрипцию мРНК с вирусной ДНК под контролем сильного промотора, локализованного в 5'-LTR.
6. Трансляцию белков Gag, Pol и Env в цитоплазме.
7. Образование вирусного капсида и упаковку в него двух РНК-цепей и молекул обратной транскриптазы.
8. Высвобождение вирионов из клетки.
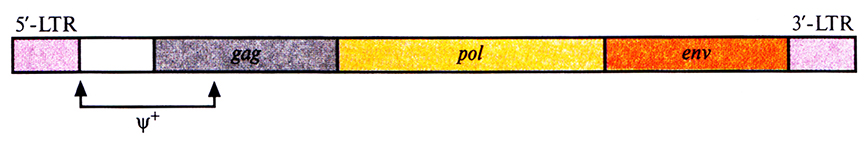
Рис. 21.2. Генетическая карта типичного ретровируса. ψ+ – последовательность, ответственная за упаковку, gag, pol и env – области, кодирующие соответственно белок капсида, белок, обладающий активностью обратной транскриптазы и интегразы, и белок оболочки. 5'-LTR содержит сигналы инициации транскрипции, причем весь геном транскрибируется как одна молекула РНК. З'-LTR содержит сигнал полиаденилирования.
Для получения ретровирусного вектора полноразмерную ДНК ретровируса встраивают в плазмиду, с помощью эндонуклеазного расщепления удаляют большую часть гена gag и гены pol и env, оставляя 5'-концевой участок гена gag и 5'- и З'-LTR, а затем рядом с ψ+ -областью встраивают «терапевтический» ген, транскрипция которого будет контролироваться 5'-LTR-пpототором; при необходимости можно встроить и маркерный селективный ген с собственным промотором (рис. 21.3). Такая конструкция позволяет экспрессировать оба клонированных гена. На основе этой схемы созданы различные ретровирусные векторы. Максимальный размер ДНК-вставки, которую может переносить ретровирусный вектор, – примерно 8 т.п.н.
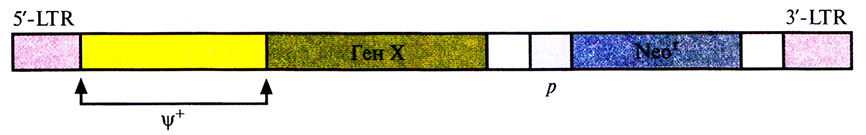
Рис. 21.3. Генетическая карта ретровирусного вектора, несущего два гена. Транскрипция «терапевтического» гена (Ген X) контролируется 5'-LTR-пpомотоpом, транскрипция селективного маркерного гена (Neor) – внутренним промотором (р). З'-LTR содержит сигнал полиаденилирования. Ψ+ – последовательность, ответственная за упаковку.
ДНК ретровирусного вектора можно использовать для трансформации клеток саму по себе, но эффективность доставки ее в ядро и интеграции в геном клетки-хозяина крайне низка. Поэтому была разработана методика упаковки полноразмерной РНК ретровирусного вектора в интактные вирусные частицы, с высокой частотой проникающие в клетку, что гарантирует встраивание соответствующей ей ДНК в геном клетки-хозяина. Для этого с помощью генной инженерии была создана «пакующая» клеточная линия, в одном из участков генома которой содержится ∆ψ+-сегмент 5'-LTR-gag-3'-LTR (т.е. сегмент, лишенный функциональной ψ+-последовательности), а в другом – ∆ψ+-сегмент 5'-LTR-pol-env-З'-LTR. Оба этих сегмента транскрибируются, но из-за отсутствия ψ+-последовательности и образования вирусных молекул РНК меньшего, чем в норме, размера формируются пустые вирусные частицы. При трансфекнии ДНК ретровирусного вектора в такие клетки она встраивается в хромосомную ДНК и транскрибируется с образованием полноразмерной РНК ретровируса, содержащей ψ+-последовательность. В таких условиях в вирусные капсиды упаковывается только РНК вектора. Образующиеся интактные вирусные частицы можно использовать для высокоэффективной доставки ретровирусного вектора в клетки-мишени (рис. 21.4).
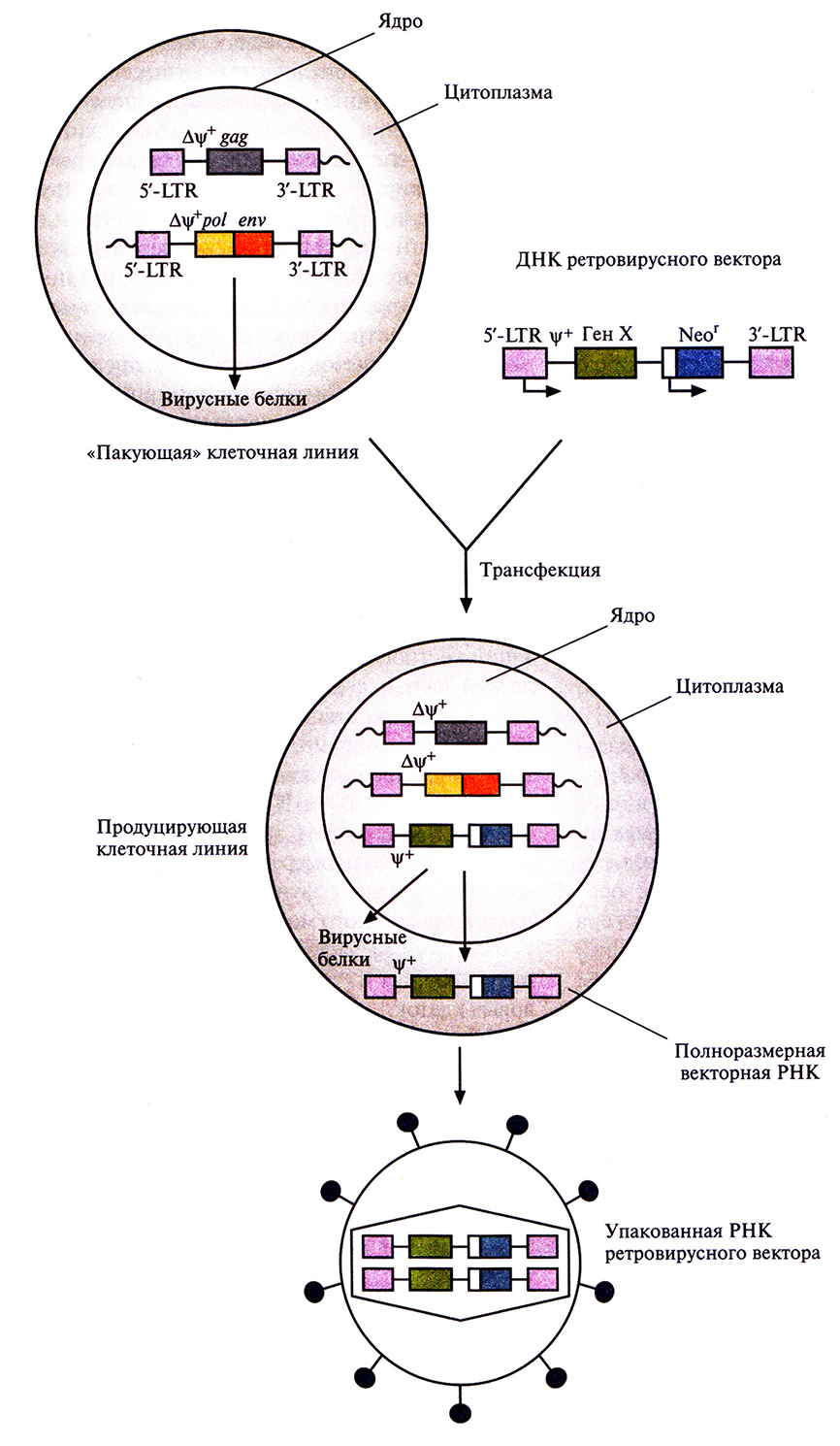
Рис. 21.4. Получение упакованного ретровирусного вектора. В двух разных участках хромосом «пакующей» клеточной линии содержатся ретровирусные гены: в одном – gag, в другом – pol и env. Их транскрипция контролируется 5'-LTR-пpомотором; оба участка лишены последовательности, необходимой для упаковки (∆ψ+). «Пакующая» линия клеток синтезирует вирусные белки, но из-за отсутствия в любой из ретровирусных мРНК последовательности, ответственной за упаковку, образуются пустые вирусные капсиды. После трансфекции «пакующей» линии клеток ретровирус ным вектором, содержащим необходимую для упаковки последовательность (∆ψ+), его полноразмерные РНК транскрибируются и упаковываются в капсиды. Высвобождаемые вирусные частицы не способны реплицироваться и в данном случае содержат «терапевтический» ген (Ген X) и селективный маркерный ген (Neor).
В «пакующей» клеточной линии не образуются компетентные по репликации ретровирусы дикого типа, способные встраиваться в гены и приводить к некотролируемой пролиферации некоторых клеток (т.е. к превращению их в раковые клетки). Это весьма существенно, особенно если частицы ретровирусного вектора предполагается использовать для геной терапии соматических клеток человека. В качестве меры предосторожности все же проводят регулярное тестирование готовых ретровирусных векторов, с тем чтобы выявить ретровирусы дикого типа. Кроме того, в «пакующей» клеточной линии нуклеотидные последовательности ретровируса и вектора локализованы в трех разных областях хромосомы, что делает весьма маловероятной возможность последовательных рекомбинационных событий, которые могли бы привести к образованию компетентного по репликации ретровируса.
Ретровирусы активно инфицируют реплицирующиеся клетки. Для переноса генов в интенсивно растущие клетки-мишени последние обрабатывают очищенными частицами упакованного ретровирусного вектора либо проводят их совместное культивирование с производящей его клеточной линией, а затем осуществляют дифференциальную селекцию для разделения клеток-мишеней и «пакующих» клеток. Трансдуцированные клетки-мишени (те, в которые при помощи вируса была перенесена невирусная ДНК) тестируют, чтобы удостовериться, что: 1) в них синтезируется продукт терапевтического гена; 2) не образуются компетентные по репликации ретровирусы; 3) ДНК ретровирусного вектора не встроилась в сайт, изменяющий способность клеток к росту либо препятствующий их нормальному функционированию. После тестирования трансдуцированные клетки наращивают в больших количествах и с разными интервалами вводят пациенту.
Наиболее вероятными кандидатами для проведения генной терапии exvivo (рис. 21.5) являются пациенты с наследственными заболеваниями, для лечения которых применяют трансплантацию костного мозга. Терапевтический эффект трансплантации костного мозга в отношении целого ряда болезней связан с наличием в нем тотипотентных эмбриональных стволовых клеток, которые встречаются с частотой 10–4–10–5, могут пролиферировать и дифференцироваться в различные типы клеток, такие как В- и Т-лимфоциты (В-клетки и Т-клетки), макрофаги, эритроциты, тромбоциты и остеокласты. Например, в том случае, когда генная мутация нарушает функции макрофагов, трансплантация костного мозга обеспечивает реципиенту постоянный запас компетентных макрофагов, происходящих из популяции тотипотентных стволовых клеток.

Рис. 21.5. Наследственные заболевания, для лечения которых применяют трансплантацию костного мозга.
Генноинженерная модификация тотипотентных стволовых клеток с их последующей инфузией или трансплантацией пациенту для замещения утраченного типа клеток или генного продукта может стать основным способом генной терапии ex vivo. В качестве примера можно рассмотреть дефект АДА, приводящий к повышению в крови уровня аденозина и дезоксиаденозина, токсическое действие которых приводит к гибели В- и Т-лимфоцитов и развитию тяжелого иммунодефицита. Поскольку и В-, и Т-лимфоциты происходят из тотипотентных стволовых клеток, перенос в последние функционального гена АДА с последующим введением их пациенту способствует понижению в крови уровней аденозина и дезоксиаденозина и предотвращает разрушение В- и Т-димфоцитов. Стволовые клетки можно получить от самого пациента (оптимальный вариант) или от совместимых доноров, которых обычно нелегко найти.
К сожалению, тотипотентные стволовые клетки очень трудно выделять из костного мозга и культивировать. Исследования по ех vivo-генной терапии SCID, вызванного дефицитом АДА, проводили на аутологичных Т-клетках, модифицированных с помощью ретровирусных векторов. Т-лимфоциты имеют ограниченное время жизни, поэтому необходимо проводить их повторные инфузии. В первом испытании двум девочкам вливали с интервалом в несколько месяцев собственные генетически «исправленные» Т-клетки, продуцирующие АДА. Наблюдаемый положительный эффект, возможно, объяснялся снижением уровня аденозина и дезоксиаденозина в крови и предотвращением гибели В- и Т-клеток.
Т клетки – не оптимальная система доставки, применяемая при генной терапии заболеваний гемопоэтических (происходящих из костного мозга) клеток. Предпочтительнее (хотя это не всегда возможно) использовать пуповинную кровь, содержащую стволовые клетки. Так, при однократном введении новорожденным с дефицитом АДА CD34+-клеток, полученных из их пуповинной крови и трансдуцированных кДНК АДА, эта кДНК поддерживалась и экспрессировалась в клетках крови неэритроидного ряда как минимум 18 мес.
В качестве примера успешной генной терапии ex vivo с использованием аутологичных клеток можно привести случай с пациенткой, гомозиготной по рецессивному гену семейной гиперхолестеролемии. Ее гепатоциты были лишены рецепторов липопротеинов низкой плотности (ЛПНП) и не разрушали холестерол; он постоянно циркулировал в крови, приводя к закупорке артерий и тяжелой болезни сердца. Лекарственные средства в подобных случаях неэффективны, а шунтирование коронарных артерий дает непродолжительный эффект.
В описываемом случае пациентке удалили около 15% печени, гепатоциты поместили в культуральную среду и ввели в них ДНК рекомбинантного ретровируса, содержащую кДНК ЛПНП-рецептора. Затем осуществили инфузию трансдуцированных гепатопитов в печень пациентки, где они прижились, экспрессировали кДНК ЛПНП-рецептора и вырабатывали функциональный рецептор как минимум 18 мес. Если брать за основу показатели содержания липидов, то состояние пациентки значительно улучшилось. Более того, в ее организме не образовывались антитела к ЛПНП-рецептору.
Генная терапия ex vivo основана на трансплантации генетически модифицированных клеток, производящих терапевтический белок. Использование собственных клеток пациента предотвращает их отторжение, но ограничивает сферу применения генной терапии ex vivo. Так, число клеток ткани-мишени может быть недостаточно для их извлечения и культивирования in vitro, некоторые соматические клетки неэффективно поглощают ДНК, а экспрессия клонированного гена иногда оказывается временной. Поэтому в настоящее время разрабатывают системы, которые защищают неаутологичные (ксеногенные, аллогенные) клетки от иммунного ответа и позволяют высвобождать «терапевтический» белок.
Неаутологичная генная терапия ex vivo включает выделение тканеспецифичных клеток, хорошо растущих в культуре (например, фибробластов или кератинонитов кожи, астроцитов мозга, гепатоцитов или миобластов), и их генетическую модификацию с помощью «терапевтического» гена. Рекомбинантные клетки заключают в искусственную полупроницаемую полимерную мембрану, через которую выходит рекомбинантный белок и поступают в клетки питательные вещества. Мембрана неиммуногенна и не вызывает сенсибилизации пациента и отторжения имплантированных клеток. В качестве инкапсулирующего материала используют разные полимеры: альгинат–поли-L-лизин–альгинат, полиэфир-сульфон, поли(акрилонитрил-ко-винилхлорид).
Доклинические испытания, проведенные in vitro и на модельных животных, показали, что инкапсулированные рекомбинантные клетки могут пролиферировать и длительное время производить большие количества рекомбинантного белка. Чтобы попытаться предотвратить гибель мотонейронов, приводящую к нейродегенеративному заболеванию боковому амиотрофическому склерозу, была проведена I фаза клинических испытаний с использованием инкапсулированных клеток, вырабатывающих цилиарный нейротрофический фактор (CNTF, от англ. ciliary neurotrophic factor). Процедуа была признана безопасной, но у пациентов с боковымамиотрофическим склерозом улучшения состояния не наблюдалось. При имплантации инкапсулированных CNTF-производящих клеток в мозг животных с химически индуцированным повреждением, моделирующим другое фатальное нейродегенеративное заболевание, хорею Ген-тингтона, клетки мозга не разрушались. Метод инкапсуляции клеток, модифицированных с помощью генной инженерии, находится на ранней стадии развития, но может стать эффективным способом доставки терапевтических генных продуктов при лечении многих заболеваний.
ВАЖНАЯ ВЕХА
Создание «пакующей» клеточной линии и ее использование для производства хелпер-независимого дефектного ретровируса
R. Mann, R. С. Mulligan, D. Baltimore Cell 33: 153–159, 1983
Вопрос о применении генои терапии всегда вызывал споры, но большинство ученых согласны с Т. Фридманом и Р. Роблином, которые еще в 1972 г. (Science 175: 949-955) писали: «Мы считаем, что геная терапия сможет облегчить симптомы некоторых наследственных заболеваний человека, поэтому необходимо продолжать исследования, направленные на ее развитие». Ключевые моменты генной терапии – адресная доставка «терапевтического» гена и обеспечение его экспрессии в определенных клетках или тканях. Сначала в качестве основного средства доставки «терапевтических» генов рассматривали векторы, полученные на основе вирусов человека. Это было связано с наличием у них механизмов проникновения в специфические клетки. Наиболее многообещающими считали векторы на основе ретровирусов. Но нативный ретровирус – это инфекционный агент, который повреждает клетки и нередко приводит к их злокачественной трансформации. Наиболее значимым достижением на пути практической реализации генной терапии стала разработка системы для упаковки «терапевтических» генов в инфекционные вирусные частицы, сохранившие способность прикрепляться к клеткам-хозяевам.
Манн и др. сконструировали первую клеточную линию для упаковки ретровирусов. Для этого они встроили вирусный геном с предварительно удаленной нуклеотидной последовательностью, ответственной за упаковку, в хромосому клеточной линии, которая в этих условиях производит неинфекционные вирусные частицы. После трансфекции этих клеток ДНК-конструкцией, которая содержала последовательность, необходимую для упаковки, и терапевтический ген, но не содержала ретровирусные гены, она упаковывалась в вирусные частицы, которые затем использовали для доставки «терапевтического» гена в нужные клетки. Этот подход был принят на вооружение другими исследователями, занимающимися генной терапией. Спустя несколько лет исходную «пакующую» клеточную линию адаптировали и для других вирусных векторов.
В 1985 г., имея в виду результаты Манна и др., У. Андерсон отметил: «В последнее время появились эффективные системы доставк-экспрессии, которые позволяют сделать генную терапию реальностью». А 22 мая 1989 г. Андерсон и его коллеги начали первое клиническое испытание по генной терапии. Предпринимавшиеся до недавнего времени попытки генной терапии не отличались особым успехом, но полученная информация свидетельствует о том, что в будущем она станет вполне обычным методом лечения многих заболеваний.
ГЕННАЯ ТЕРАПИЯ IN VIVO
Генная терапия in vivo предполагает доставку «терапевтического» гена непосредственно в клетки определенной ткани пациента (рис. 21.6). Ретровирусные векторы проникают только в делящиеся клетки-мишени, однаково многих тканях, на которые направлена генная терапия, большинство клеток не делится. Поэтому были разработаны разнообразные вирусные и невирусные векторные системы доставки «терапевтических» генов, учитывающие большое число потенциальных тканей-мишеней (кожа, мышцы, легкие, мозг, толстая кишка, селезенка, печень, клетки крови) и расположение их в организме человека. «Идеальная» система доставки должна обеспечивать высокую эффективность поглощения «терапевтического» гена клетками-мишенями, минимальность его внутриклеточного разрушения при транспорте в ядро и поддержание уровня экспрессии, достаточного для облегчения состояния больного.
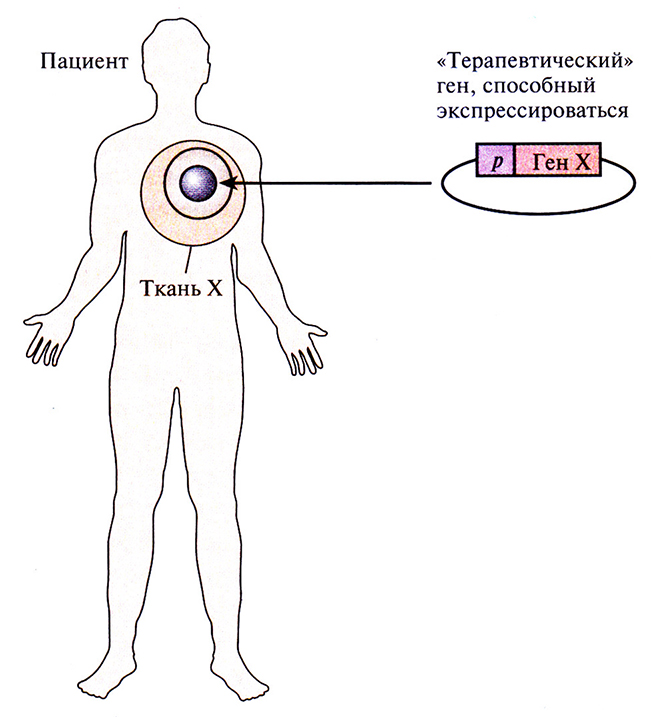
Рис. 21.6. Схематическое представление генной терапии in vivo. Клонированный «терапевтический» ген (Ген X) кодирует белок, корректирующий генетический дефект. Этот ген доставляется к клеткам определенной ткани пациента с наследственным заболеванием и экспрессируется в них. Промотор р, под контролем которого осуществляется транскрипция, тканеспецифичен.
ВИРУСНЫЕ СИСТЕМЫ ДОСТАВКИ ГЕНОВ
Ретровирусные векторы
Опыт клинических испытаний с участием более 200 пациентов показывает, что дефектные по репликации ретровирусные векторы не оказывают каких-либо неблагоприятных побочных эффектов. Тем не менее безопасности их применения продолжают придавать большое значение. Создана конструкция, называемая плазмовирусом, которая содержит ретровирусные гены gag и poh находящиеся под контролем 5'-LTR-npомотоpa, а также «терапевтический» ген и ген env, управляемые цитомегаловирусным промотором. После трансфекции плазмовирус запускает образование дефектных по репликации вирусных частиц, причем вероятность рекомбинации с образованием компетентных по репликации ретровирусов очень мала. Вектор может переносить не более 3,5 т. п. н. ДНК, но и длина большинства потенциальных «терапевтических» кДНК и генов – супрессоров опухолей составляет 0,5–2 т.п.н.
В ретровирусную векторную систему внесены дополнительные усовершенствования: увеличено число образующихся вирусных частиц, повышена эффективность трансдукции, осуществлена генноинженерная модификация, обеспечивающая их проникновение в неделяшиеся клетки, повышена специфичность инфекции. В последнем случае геном рекомбинантного ретровирусного вектора упаковывается в оболочку другого вируса, белок которой и определяет специфичность связывания ретровируса и спектринфицируемых им клеток. Это явление называется фенотипическим смешиванием (pseudotype formation). Фенотипически смешанный вирус получают с помощью котрансфекции клеточной линии, которая синтезирует продукты генов gag и pol, рекомбинантным ретровирусным вектором и вектором, экспрессирующим ген env другого вируса. Изменяя ген env, можно как сузить спектр инфицируемых вирусом клеток до строго определенного типа, так и расширить его. Кроме того, в ген env ретровируса можно встроить нуклеотидную последовательность, кодирующую пептид, который связывается с определенным клеточным рецептором и обеспечивает внедрение рекомбинантного ретровируса в нужные клетки. И наконец, можно добиться специфичности экспрессии терапевтического гена, осуществляя ее под контролем промотора, специфичного для определенных клеток.
Аденовирусные векторы
Аденовирусы инфицируют неделящиеся клетки человека и широко используются в качестве живых вакцин, которые предотвращают респираторные инфекции и гастроэнтериты, не оказывая побочного действия. Эти свойства делают аденовирусы перспективными для доставки генов в клетки-мишени.
Для получения аденовирусного вектора провели котрансфекцию клеточной линии, синтезирующей продукты аденовирусного гена Е1, двумя участками генома аденовируса (рис. 21.7). Один из них может существовать в виде плазмиды в Е. coli и содержит вместо Е1-области «терапевтический» ген, фланкируемый нуклеотидными последовательностями аденовируса, а второй представляет собой молекулу ДНК аденовируса, которая лишена 5'-концевого участка, включающего El-область, и имеет перекрывающийся участок с несущей терапевтический ген плазмидой. Рекомбинация между двумя трансфицирующими фрагментами ДНК в области их перекрывания приводит к восстановлению полноразмерного аденовирусного гена, в котором вместо Е1 -области находится терапевтический ген. Продукты гена Е1, поставляемые клеткой-хозяином, инициируют образование вирусных частиц, высвобождающихся из клетки в результате лизиса. Клонирующая емкость аденовирусного вектора составляет около 7,5 т.п.н. В отсутствие рекомбинации трансфицирующие молекулы ДНК, обладающие недостаточной длиной, не могут упаковываться в вирусные частицы. Вероятность того, что между областью Е1 в геноме клетки-хозяина и ДНК рекомбинантного аденовируса произойдет рекомбинация с образованием компетентных по репликации вирусов, чрезвычайно мала.
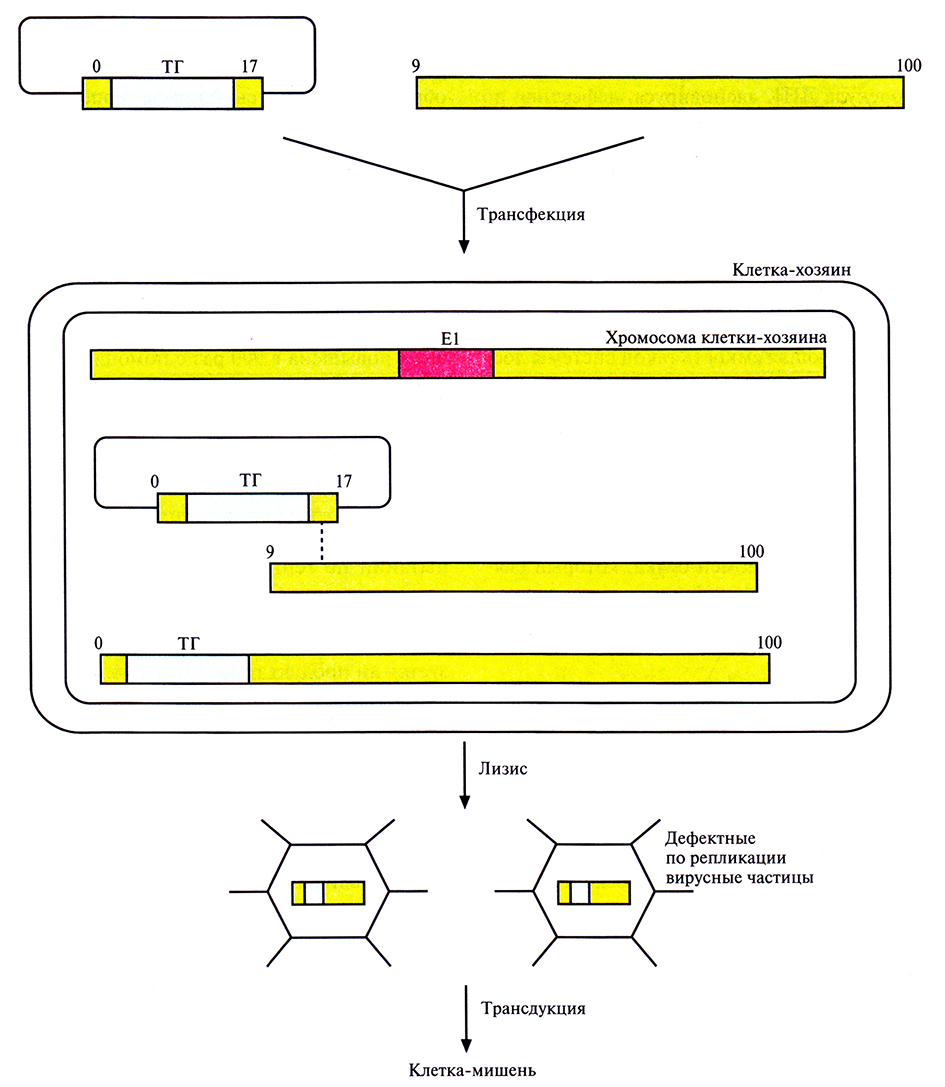
Рис. 21.7. Аденовирусный вектор. В клетку-хозяина, несущую интегрированный в геномную ДНК функциональный ген Е1 аденовируса, вводят встроенную в сегмент аденовирусного генома (0–17 единицы карты) плазмиду с «терапевтическим» геном (ТГ) и участок геномной ДНК аденовируса (9–100 единицы карты). Длина генома аденовируса равна 100 единицам. В результате рекомбинации (штриховая линия) между перекрывающимися участками плазмиды и ДНК аденовируса образуется молекула ДНК, эквивалентная полноразмерному вирусному геному. Рекомбинантная ДНК, содержащая «терапевтический» ген упаковывается и высвобождается из клетки после лизиса. Образующиеся вирусные частицы дефектны по репликации. Плазмидная ДНК, входящая в состав конечной генетической конструкции, не влияет на упаковку рекомбинантной ДНК (не показано).
После того как рекомбинантный аденовирус инфицирует клетку-мишень, его ДНК проникает в ядро, где и происходит экспрессия «терапевтического» гена. Рекомбинантная ДНК. не интегрирует в хромосому и сохраняется непродолжительное время, поэтому при проведении генотерапии с использованием аденовирусных векторов необходимо вводить их с определенной периодичностью.
Аденовирусные векторы использовали в клинических испытаниях по генной терапии муковисцидоза. Первые результаты не обнадеживали: ген трансмембранного регуляторного белка, нарушения в котором приводят к муковисцидозу (CFTR, от англ. cystic fibrosis transmembrane regulator), был перенесен лишь в небольшое число клеток пациента, а многократное введение рекомбинантного аденовируса и низкий уровень экспрессии некоторых аденовирусных генов привели к развитию у пациентов выраженного иммунного ответа и гибели трансдуцированных клеток.
Эту проблему решали разными путями. Например, сконструировали «пакующую» клеточную линию, содержащую El-область и ряд аденовирусных генов, которые не входят в состав трансдуцирующей ДНК и не попадают в клетки-мишени. Затем удалось добиться того, чтобы ни один из аденовирусных генов не включался в трансдуцирующую ДНК. Для этого линеаризовали плазмиду Е. coli (28 т.п.н.), которая обеспечивает экспрессию одного или большего числа терапевтических генов и не содержит аденовирусных генов, и пришили к ее концам фрагменты ДНК (по 4 т. п. н.), содержащие точку начала репликации аденовирусной ДНК, последовательность, ответственную за ее упаковку, и сигнал терминации. Длина продукта лигирования (36 т.п.н.) соответствует длине полноразмерного генома аденовируса. Затем полученным продуктом и аденовирусным геномом без El-области и последовательности, ответственной за упаковку, провели котрансфекцию клетки-хозяина, экспрессирующей El-гены. Молекула ДНК аденовируса, дефектная по репликации и упаковке, поставляет гены для синтеза компонентов вируса, а продукт дотирования реплицируется и упаковывается в вирусные частицы. При этом около 99% высвобождаемых вирусных частиц содержат молекулу ДНК с «терапевтическим» геном (генами). С помощью центрифугирования их можно отделить от дефектных по репликации вирусов, которые все же образуются в незначительном количестве. ДНК-клонирующая емкость такой системы достигает 28 т.п.н.
Эффекта вность аденовирус-опосредованно го переноса генов можно повысить, если сконструировать вирус, проникающий преимущественно в определенную клетку-мишень. Для этого в ген, ответственный за образование нитей аденовируса, следует включить последовательность, кодирующую домен белка, который связывается с клеточноспецифичным рецептором.
Векторы на основе аденоассоциированных вирусов
Дденоассоциированные вирусы (ААВ) – это небольшие непатогенные вирусы человека с одноцепочечным ДНК-геномом (4,7 т.п.н.), который может интегрировать в специфический сайт 19-й хромосомы. Такое название они получили потому, что для продуктивной инфекции им необходимы белки другого вируса (вируса-помощника), например аденовируса. После того как ААВ попадает в ядро, его геном с помощью полимераз клетки-хозяина преобразуется в двух-цепочечную ДНК и транскрибируется.
Отсутствие патогенности делает ААВ весьма перспективным вектором для доставки в организм человека «терапевтических» генов. Рекомбинантный ААВ получают с помощью котрансфекции клетки-хозяина, инфицированной каким-нибудь аденовирусом (вирусом-помощником), двумя плазмидами (рис. 21.8). Одна из них несет «терапевтический» ген, фланкированный инвертированными концевыми повторами (длиной от 125 п. н.) ААВ, а вторая – два его гена, rep и cap, ответственные за репликацию генома и синтез капсида соответственно. После лизиса инфицированных клеток рекомбинантные ААВ отделяют от аденовируса с помощью центрифугирования и диализа, а оставшиеся в образце аденовирусы (вирусы герпеса) инактивируют нагреванием. Рекомбинантный ААВ может нести ДНК-вставку размером до 4,5 т.п.н., не вызывает развития иммунного ответа, поскольку не содержит ААВ-генов, но и не может интегрировать в 19-ю хромосому из-за отсутствия гена rep.
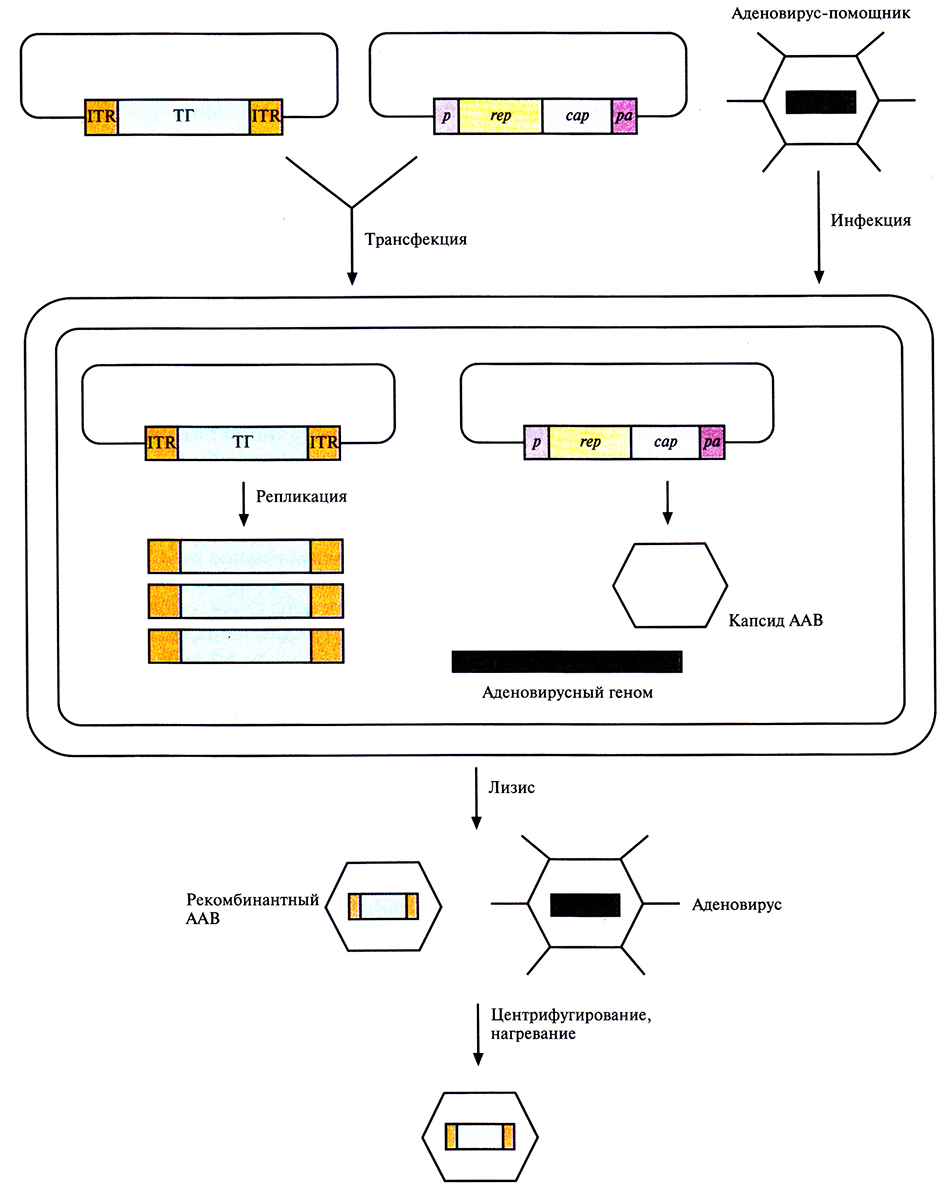
Рис. 21.8. Вектор на основе аденоассоциированного вируса (ААВ). Проведена котрансфекция клетки-хозяина, инфицированной аденовирусом-помощником, двумя плазмидами, одна из которых содержит «терапевтический» ген (ТГ), фланкированный инвертированными концевыми повторами (ITR) ААВ, а другая – гены ААВ, ответственные за репликацию (rep) и формирование капсида (cap), которые находятся под контролем промотора (р), и последовательность полиаденилирования (ра). Высвободившиеся после лизиса частицы рекомбинантного ААВ и аденовируса разделяют центрифугированием, а оставшиеся аденовирусные частицы инактивируют нагреванием.
В одном из доклинических испытаний эффективность in vivo трансдукции гепатоцитов мыши (рекомбинантный ААВ вводили внутривенно) была повышена в 900 раз с помощью предварительного облучения печени нетоксическими дозами и введения нерекомбинантного (дикого типа) ААВ. В этом случае кДНК фактора IX системы свертывания крови («терапевтический» ген) экспрессировалась в течение как минимум 5 мес на уровне, достаточном для коррекции дефекта при гемофилии. В I фазе клинических испытаний по генной терапии муковисцидоза в легкие вводили CFTR-AAВ-вектор; при этом не развивалась воспалительная реакция, а вектор сохранялся до 70 сут. Чтобы определить, образуется ли продукт гена CFTR в количестве, достаточном для достижения терапевтического эффекта, нужны дальнейшие клинические испытания.
Векторы на основе вируса простого герпеса
Для того чтобы ретро- и аденовирусные векторы инфицировали специфические типы клеток, нужно модифицировать их с помощью генной инженерии, однако в природе существуют вирусы, уже обладающие сродством к определенному типу клеток. Так, вирус простого герпеса 1 типа (HSV) инфицирует нейроны и персистирует в них, часто вызывая у человека так называемые «простудные» высыпания, а иногда – энцефалит с летальным исходом. Вирус присутствует в нейронах в латентной форме, а при стрессе и гормональных нарушениях инициируется литический цикл.
Существует множество заболеваний, поражающих центральную и периферическую нервную систему: опухоли, метаболические и иммунные нарушения, нейродегенеративные заболевания (болезнь Альцгеймера, болезнь Паркинсона). Неврологические заболевания, как правило, бывают хроническими и приводят к госпитализации больного чаще, чем все остальные болезни вместе взятые. Вследствие тропности HSV к нервным клеткам он является подходяшим вектором для генной терапии таких заболеваний.
Геном HSV представляет собой двухцепочечную молекулу ДНК длиной 152 т. п. н. Капсид вируса сливается с мембраной нейрона, и его ДНК транспортируется в ядро. Репродуктивный цикл вируса состоит из литической (репликация ДНК и образование вирусных частиц) и латентной (конденсация вирусного генома и активация как минимум двух так называемых латентно-ассоциированных промоторов) фаз.
Замена сегмента геномаддиной примерно 30 т.п.н. ДНК-вставкой не оказывает заметного влияния на репликацию HSV, его упаковку или инвазионную способность. С другой стороны, большой размер генома HSV затрудняет генетические манипуляции с ним. Для решения этой проблемы в плазмиду Е. coli, которая может переносить до 8 т.п.н. чужеродной ДНК, встроили «усеченный» геном HSV, состоящий из точки инициации репликации и последовательности, ответственной за упаковку. Полученные HSV-производные назвали ампликонами (ампликон-плазмидами).
Большинство систем доставки генов на основе HSV предполагает использование вируса-помощника, который поставляет белки, необходимые для репликации и сборки вируса, но не образует инфекционные вирусные частицы, поскольку его геном модифицирован и не способен упаковываться. Для получения рекомбинантного HSV осуществляют трансфекцию ампликон-плазмиды в инфицированную вирусом-помощником клетку-хозяина. ДНК ампликона реплицируется по типу «катящегося кольца»: внутренняя кольцевая цепь играет роль матрицы, а во внешней происходит разрыв, и к свободной 3'-концевой ОН-группе ковалентно присоединяются нуклеотиды. Растущая цепь представляет собой линейную тандемную последовательность сегментов, комплементарных внутренней цепи, и, отсоединяясь от нее, сама становится матрицей для синтеза комплементарной цепи. В результате образуется линейная двухцепочечная молекула – множественная копия ампликона. Длина каждого ампликона составляет 15 т. п. н., поэтому набор из 10 тандемных копий соответствует полноразмерному геному HSV и упаковывается в HSV-капсид (рис. 21.9).
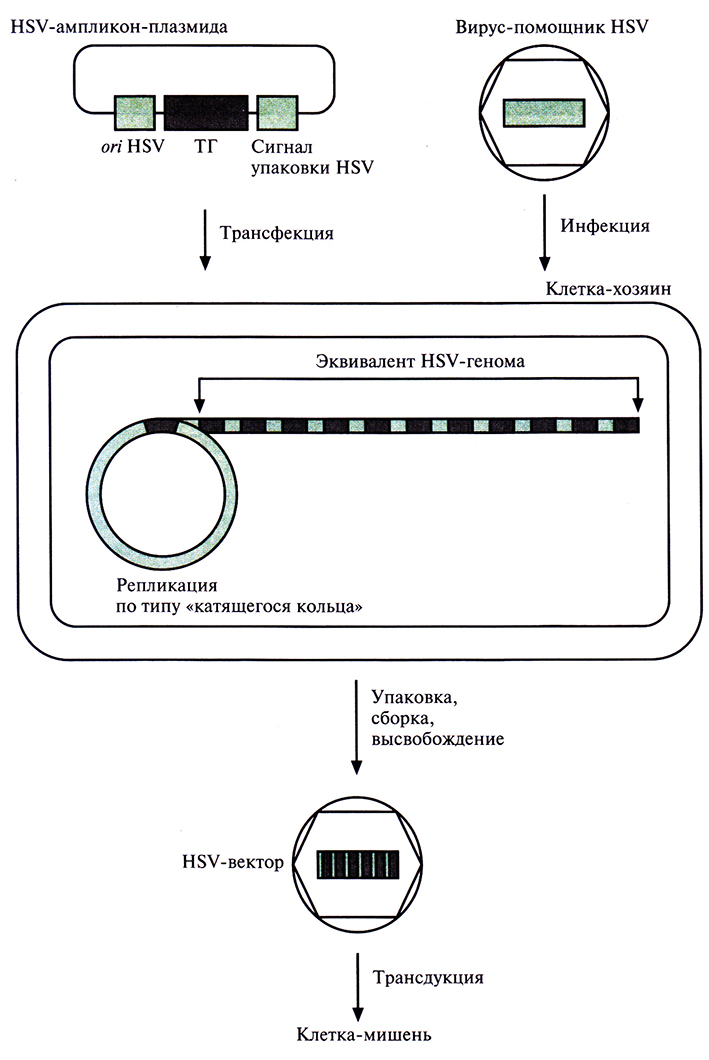
Рис. 21.9. Вектор на основе HSV-ампликон-плазмиды. Точка инициации репликации HSV (on HSV), сигнал упаковки HSV и «терапевтический» ген (ТГ) встраивают в плазмиду Е. coli (HSV-ампликон-плазмида). Проводят трансфекцию клетки-хозяина, инфицированной вирусом-помощником HSV, полученной плазмидой. ДНК ампликон-плазмиды реплицируется по типу «катящегося кольца». 10 ампликонов, соответствующих полноразмерному геному HSV, упаковываются в HSV-капсид, который поставляет вирус-помощник HSV. Геном этого вируса не упаковывается. HSV-частицы, несущие множество копий «терапевтического» гена, высвобождаются при лизисе клетки и используются для трансдукции нейронов.
Рекомбинантный HSV можно получить и с помощью котрансфекции клеток-хозяев, в которых вирус может реплицироваться, с помощью ДНК HSV дикого типа и плазмиды, которая содержит «терапевтический» ген, фланкированный последовательностями ДНК из вспомогательных участков HSV-генома. ДНК HSV дикого типа реплицируется в ядре клетки-хозяина, при этом в результате рекомбинации «терапевтический» ген может встроиться в HSV-reном. Затем частицы как рекомбинантного, так и дикого типа HSV упаковываются и высвобождаются из клеток. Доля рекомбинантных HSV в общем вирусном пуле очень мала, поэтому вирусы размножают, а затем с помощью ПЦР или гибридизации выявляют «терапевтический» ген в образовавшихся бляшках. Рекомбинантный вирус хранят в условиях, не допускающих его загрязнения HSV дикого типа (рис. 21.10).
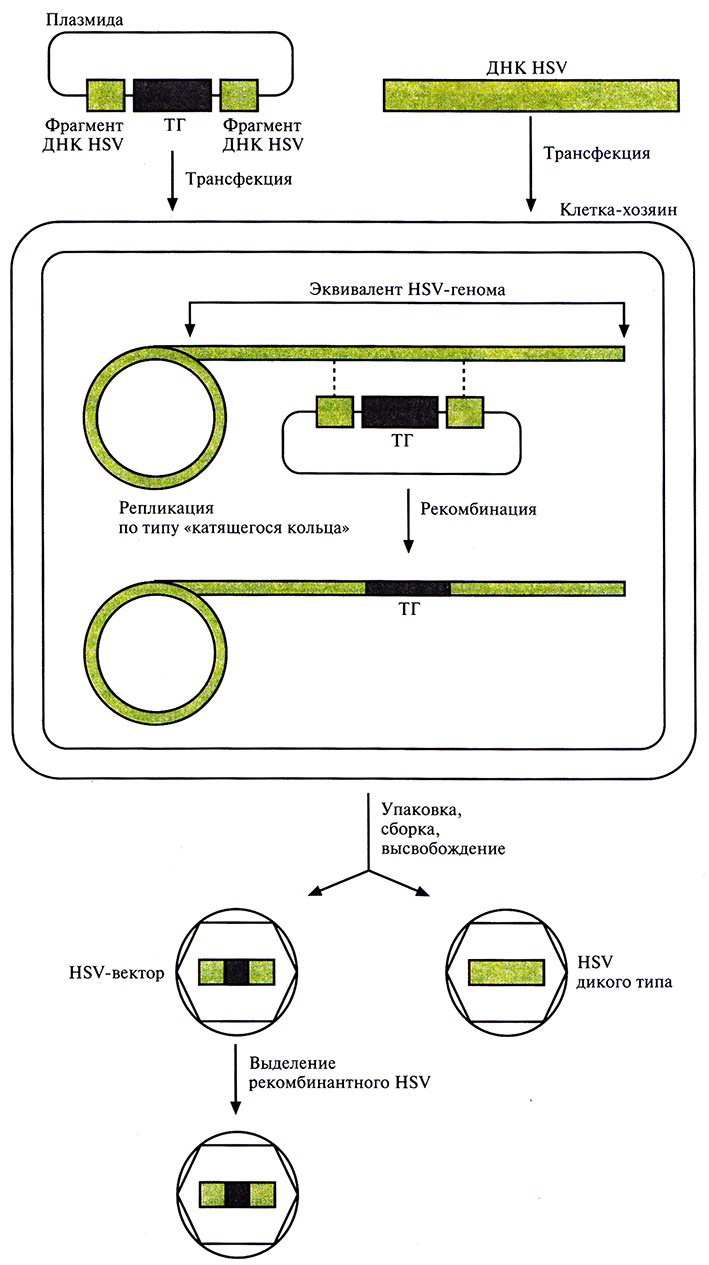
Рис. 21.10. Образование HSV-вектора с помощью рекомбинации. Проводят котрансфекцию клетки-хозяина плазмидой, которая содержит «терапевтический» ген, фланкированный последовательностями ДНК из вспомогательных областей HSV-генома, и ДНК HSV дикого типа. HSV-геном реплицируется в клеточном ядре по типу «катящегося кольца», при этом между фрагментами ДНК HSV, входящими в состав плазмиды, и ДНК HSV дикого типа может произойти рекомбинация (штриховая линия). Молекулы ДНК HSV дикого типа и рекомбинантного HSV упаковываются в вирусные частицы, высвобождающиеся из клетки после лизиса. Вирусы размножают и проводят скрининг бляшек для идентификации рекомбинантных HSV. Полученные HSV-векторы хранят в условиях, исключающих их загрязнение HSV дикого типа.
Доклинические испытания на экспериментальных животных показали, что гены, доставленные с помощью HSV-векторов в клетки мозга и периферической нервной системы, экспрессируются и поддерживаются длительное время. Однако до начала I фазы клинических испытаний HSV-векторов необходимо провести дополнительные исследования.
НЕВИРУСНЫЕ СИСТЕМЫ ДОСТАВКИ ГЕНОВ
В опосредованной вирусами доставке генов участвуют клеточные рецепторы, с помощью которых вектор проникает в клетку-мишень, не разрушаясь лизосомными ферментами, и векторная ДНК попадает в ядро. Однако вирусные векторы имеют ряд недостатков: они дорогостоящи и часто обладают ограниченной клонирующей емкостью, что не позволяет регулировать экспрессию «терапевтического» гена с помощью тканеспецифичных последовательностей. Кроме того, вирусные белки могут вызывать воспалительную реакцию, что исключает повторное введение вектора. Поэтому были разработаны невирусные системы доставки генов.
Самая простая из них – прямое введение ДНК-конструкций в клетки ткани-мишени. Если в скелетную мышцу мыши инъецировать плазмидную ДНК, то она проникнет в некоторое число клеток, о чем свидетельствует экспрессия гена-репортера в течение по крайней мере 50 сут. Однако применение этого подхода ограничивается тем, что не все ткани доступны для инъекций, а кроме того, нужны большие количества ДНК Можно бомбардировать с помощью генного «ружья» клетки кожи или – через надрез – клетки подкожной опухоли конъюгированными с ДНК частицами золота диаметром 1–3 мкм. Введенные таким образом терапевтические» гены экспрессируются в тканях-мишенях, а их продукты поступают в кровь. Это может облегчить доставку терапевтического белка в ткань-мишень, прямой доступ к которой затруднен. Однако большинство белков, в норме не присутствующих в крови, инактивируются или разрушаются ее компнентами. Для решения этой проблемы нужны дополнительные исследования.
Проникновение ДНК через клеточную мембрану можно облегчить, окружив генетическую конструкцию искусственной липидной оболочкой, образующей липидную сферу (липосому) с водным содержимым. Созданы липосомы с самыми разными свойствами, например катионные липосомы, поверхность которых заряжена положительно; они связываются с отрицательно заряженной молекулой ДНК, образуя ДНК-липидный комплекс (липоплекс). Липоплексы легко образуются, относительно нетоксичны и неиммуногенны, но эффективность переноса генов с их помощью невысока, поскольку большая часть ДНК после попадания в клетку захватывается лизосомами и разрушается. Так, в одном из клинических испытаний по генной терапии муковисцидоза с использованием комплекса липосома– CFTR-тен частота трансфекции клеток назального эпителия оказалась довольно низкой, а экспрессия гена – непродолжительной.
Для доставки в клетки крупных генетических конструкций (>10 т.п.н.) с помощью эндосомного клеточного транспорта, позволяющего избежать лизосомного разрушения ДНК, образуют конъюгат ДНК с другими молекулами. Для этого поли-L-лизин ковалентно сшивают с молекулой, связывающейся со специфическим клеточным рецептором, а затем добавляют ДНК. В результате получается компактная, плотно скрученная структура (тор), на внешней поверхности которой располагаются сайты связывания с клеточным рецептором (рис. 21.11). К сожалению, подобный конъюгат, несмотря на свою специфичность, обладает низкой эффективностью трансфекции. Все созданные к настоящему времени невирусные системы доставки имеют два основных недостатка: 1) низкая частота трансфекции, не позволяющая достичь нужного терапевтического эффекта; 2) непродолжительное время экспрессии «терапевтического» гена, не обеспечивающее эффективного лечения.
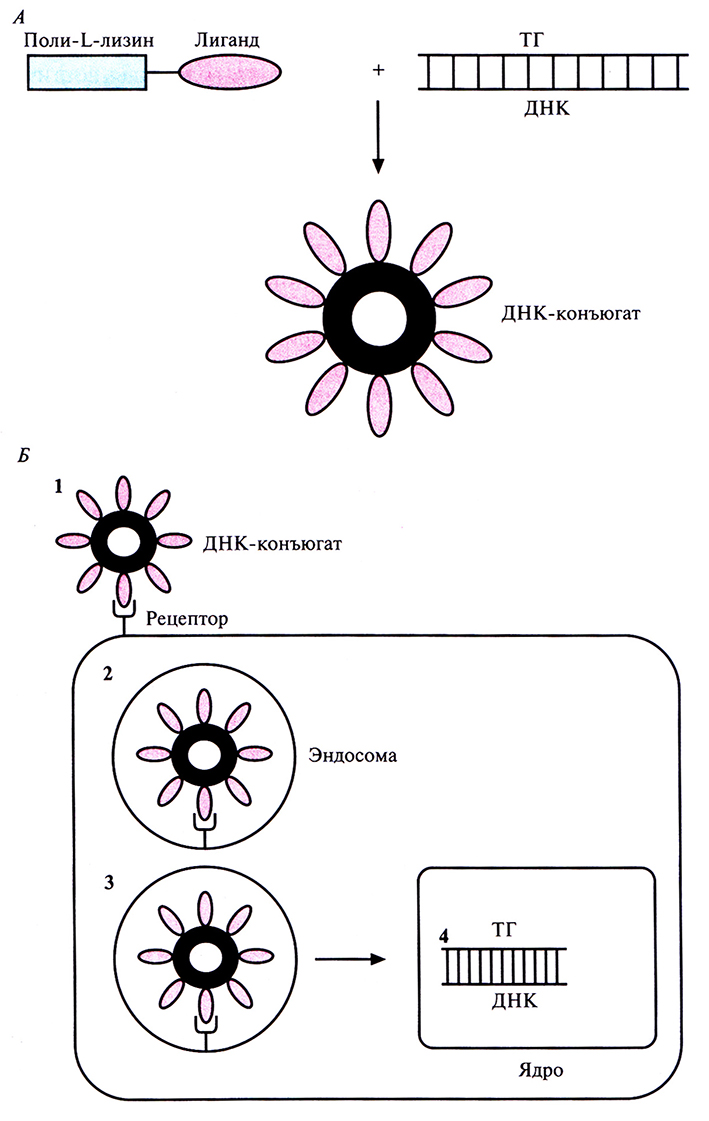
Рис. 21.11. Система доставки «терапевтических» генов с использованием ДНК-конъюгата. А. К пол и-L-лизину пришивают лиганд, соединяющийся с поверхностным клеточным рецептором, и добавляют ДНК, содержащую «терапевтический» ген. В результате образуется конденсированная структура, на поверхности которой располагаются лиганды. Б. ДНК-конъюгат связывается со специфическим клеточным рецептором (1) и обволакивается клеточной мембраной (2) с образованием эндосомы (3), которая защищает его от лизосом. В эндосоме часть молекул ДНК высвобождается из конъюгата и проникает в ядро клетки (4), где и происходит экспрессия «терапевтического» гена.
Возможно, подходящим терапевтическим вектором станет искусственная хромосома человека. Это связано с: 1) возможностью включения в нее протяженных сегментов чужеродной ДНК вместе с полным набором регуляторных элементов для одного или нескольких «терапевтических» генов; 2) возможностью использования геномного варианта «терапевтического» гена, обеспечивающего высокую эффективность его экспрессии; 3) стабильностью «терапевтического» гена и его длительной экспрессией как в пролиферирующей, так и в неделящейся клетке-мишени.
Искусственная хромосома содержит три основных элемента: концевые участки (теломеры), центромеру и точки инициации репликации. Свойства теломерных областей хромосом человека хорошо изучены, чего нельзя сказать о центромерах и точках инициации репликации, и существовали опасения, что искусственную хромосому человека не удастся сконструировать, пока не будут досконально изучены все ее элементы. Однако уже получены и поддерживаются в трансфицированной культуре клеток стабильные линейные искусственные хромосомы человека (микрохромосомы), состоящие из множества ДНК-повторов (длиной около 1 м. п. н.) центромерной области Y-хромосомы, высокомолекулярных фрагментов геномной ДНК и теломерных участков. В их центромерную область был встроен ген устойчивости к неомицину, что позволило использовать среду G418 в качестве селективной. В нескольких G418-устойчивых клетках были обнаружены микрохромосомы длиной от 6 до 10 м.п.н.
Две из трех микрохромосом были получены «усечением» существующей хромосомы. В одном случае исходная центромера была сохранена, а в другом заменена трансфицированной центромерной областью. Третью, полностью искусственную микрохромосому получили лигированием in vitro трех трансфицированных ДНК-элементов. Ясно, что создание искусственной хромосомы человека, содержащей «терапевтический^)» ген(ы), вполне реально, но основной проблемой станет доставка этой огромной молекулы ДНК в ядро клетки-мишени. Кроме того, экспрессия генов, входящих в состав ДНК-блоков, из которых построена искусственная хромосома, может оказывать вредные воздействия на клетки-мишени. Для начала в ткани пациента можно попытаться имплантировать инкапсулированные клетки с искусственными хромосомами.
АКТИВАЦИЯ ПРЕДШЕСТВЕННИКА
ЛЕКАРСТВЕННОГО ВЕЩЕСТВА («ПРОЛЕКАРСТВА»)
Несмотря на широкое применение хирургических методов лечения, лучевой и химиотерапии, злокачественные новообразования по-прежнему остаются одной из основных причин смерти людей, поэтому задача разработки новых способов их лечения является весьма актуальной. Одним из таких способов является уничтожение пролиферирующих опухолевых клеток с помощью ганцикловира [GCV; 9-(1,3-дигидрокси-2-пропоксиметил)гуанина], активированного продукта гена тимидинкиназы вируса простого герпеса (HSVtk). Для этого проводят in vivo трансдукцию или трансфекцию опухолевых клеток геном HSVtk, находящимся под контролем активного промотора, а через несколько дней вводят ганцикловир. Вирусная тимидинкиназа фосфорилирует ганцикловир с образованием ганциктовирмонофосфата. Киназы клетки-хозяина почти не фосфорилируют ганцикловир, зато охотно присоединяют фосфатные группы к его монофосфату с образованием ганцикловиртрифосфата, который ингибирует ДНК-полимеразу и останавливает синтез ДНК, вызывая гибель пролиферирующих клеток. Кроме того, через межклеточные контакты ганцикловиртрифосфат может проникать в нетранссрицированные опухолевые клетки, приводя и к их гибели. Одна экспрессирующая ген HSVtk опухолевая клетка может уничтожить до 10 немодифицированных клеток. Это явление называется «эффектом свидетеля».
Ген, вызывающий при определенных условиях гибель собственной клетки, называют геном «самоубийства», а термин «пролекарство» относится к неактивной форме лекарственного вещества, которая активируется с помощью другого компонента терапевтической системы. Разработаны и другие комбинации «пролекарство» – ген-активатор, но система GCV– HSVtk используется чаще других.
Эффективность системы GCV– HSVtk доказана целым рядом доклинических испытаний. Однако I фаза ее клинических испытаний, в которых участвовали больные с терминальной стадией рака, не показала регресса опухоли. Возможно, ген HSVtk был трансдуцирован в слишком малое число опухолевых клеток и, несмотря на «эффект свидетеля», не мог подавить рост опухоли. В настоящее время разрабатываются новые подходы, которые смогут повысить частоту транедукции и доставить ген HSVtk в клетки по всему объему опухоли.
Для генной терапии рака разработаны также комбинированные подходы, использующие две разные системы генов. В одном из них сочетаются GCV– HSVtk-терапия и генная иммунотерапия (рис. 21.12). Одну часть опухолевых клеток транедуцируют геном HSVtk, другую – клонированной кДНК (или геном) одного из цитокинов. Цитокины (интерлейкин-2, интерлейкин-12 и другие) играют роль сигнала, мобилизующего клетки иммунной системы и стимулирующего иммунный ответ. Показано, что опухолевые белки, которые высвобождаются из клетки, уничтоженной в результате терапии с помощью гена «самоубийства», взаимодействуют с иммунными клетками, привлекаемыми к месту локализации опухоли цитокином, и запускают противоопухолевую иммунную реакцию. Кроме того, противоопухолевые антитела, поступая в кровоток и циркулируя по всему организму, предотвращают появление метастазов.
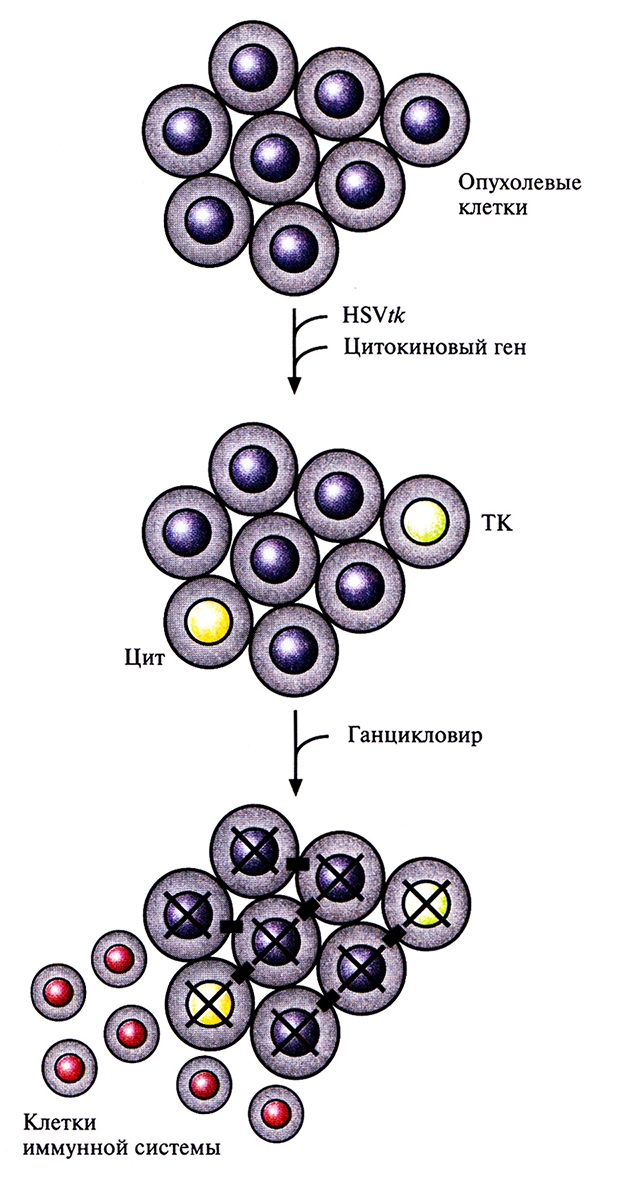
Рис. 21.12. Комбинированный подход с использованием GCV–HSVtk-терапии и генной иммунотерапии. Опухолевые клетки трансдуцировали in vivo геном тимидинкиназы вируса простого герпеса (HSVtk) и цитокиновым геном. Трансдуцированные клетки опухоли, которые синтезируют цитокин (Пит), привлекают иммунные клетки, а те, которые синтезируют тимидинкиназу (ТК), фосфорилируют ганииковир. Фосфорилированный ганцикловир проникает через межклеточные контакты (черные прямоугольники) в соседние клетки и, связываясь с ДНК, уничтожает их (X).
Этот подход к генной терапии рака был проверен экспериментально: в печень животных имплантировали клетки рака толстой кишки, раздельно транедуцированные генами HSVtk одного из цитокинов. Введение ганцикловира останавливало рост опухоли в печени. Опухоль не возникала и при введении нетранедуцированных опухолевых клеток в другие ткани такого животного. У контрольных животных в аналогичных условиях происходило развитие опухолей во всех местах введения нетранедуцированных клеток рака толстой кишки. Несмотря на столь многообещающие результаты, прежде чем приступать к клиническим испытаниям терапии с использованием гена «самоубийства» или различных комбинаций генной терапии, необходимо установить, какие опухоли будут поддаваться такому лечению и не вызовет ли оно побочных эффектов.
ЛЕКАРСТВЕННЫЕ СРЕДСТВА НА ОСНОВЕ ОЛИГОНУКЛЕОТИДОВ
В большинстве протоколов генной терапии ех vivo и in vivo используются клонированные генетические конструкции, возмещающие функциональную форму белка, который не синтезируется в организме больного или синтезируется в дефектной форме. Однако многие заболевания человека (рак, воспаления, вирусные и паразитарные инфекции) связаны, напротив, с гиперпродукцией нормального белка. Для лечения таких состояний разработаны терапевтические системы с использованием олигонуклеотидов. Небольшой олигонуклеотид может гибридизоваться со специфическим геном или мРНК и снижать уровень транскрипции или трансляции, уменьшая тем самым количество синтезируемого белка, ответственного за патологию. Олигонуклеотид, который гибридизуется с самим геном и блокирует его транскрипцию, называется «антигенным», а тот, который гибридизуется с соответствующей мРНК, – «антисмысловым». Снизить уровень транскрипции и трансляции гена-мишени может также олигонуклеотид, который связывается с фактором транскрипции, контролирующим экспрессию специфического гена. Для предотвращения активации транскрипции специфических генов можно использовать и двухцепочечные олигонуклеотиды, присоединяющиеся к ДНК-связывающим белкам. Можно создать также синтетические молекулы ДНК, которые присоединяются к специфическим белкам-мишеням, исходно не являющимся ДНК-связывающими, и тем самым блокируют их функционирование. Наконец, для уменьшения количества определенной мРНК и синтезируемого на ней белка можно модифицировать рибозимы – природные РНК-последовательности, которые связываются со специфическими молекулами РНК и разрезают их. В будущем лекарственные средства на основе нуклеиновых кислот, по-видимому, найдут широкое применение, при этом главным объектом научных исследований и клинических испытаний будут «антисмысловые» последовательности и особенно – «антисмысловые» олигонуклеотиды.
Синтез «антисмысловых» м РНК in vivo
«Антисмысловая» РНК, которую предполагается использовать в качестве лекарственного средства, должна связываться с определенной мРНК и ингибировать трансляцию кодируемого ей белка, подавляя тем самым патологический процесс (рис. 21.13). Для получения таких РНК использовали экспрессирующие векторы, несущие ДНК-вставки в такой ориентации, чтобы их транскрипты были антисмысловыми по отношению к мРНК. В качестве примера можно привести эписомные экспрессирующие векторы, в которые встроены кДНК инсулиноподобного фактора роста 1 (IGF-1) или его рецептора (IGF-1R), находящиеся в обратной ориентации под контролем металлотионеинового промотора, активируемого ZnS04. IGF-1 вырабатывается в избыточном количестве клетками злокачественной глиомы, наиболее распространенной опухоли головного мозга, a IGF-1R – клетками карциномы предстательной железы.
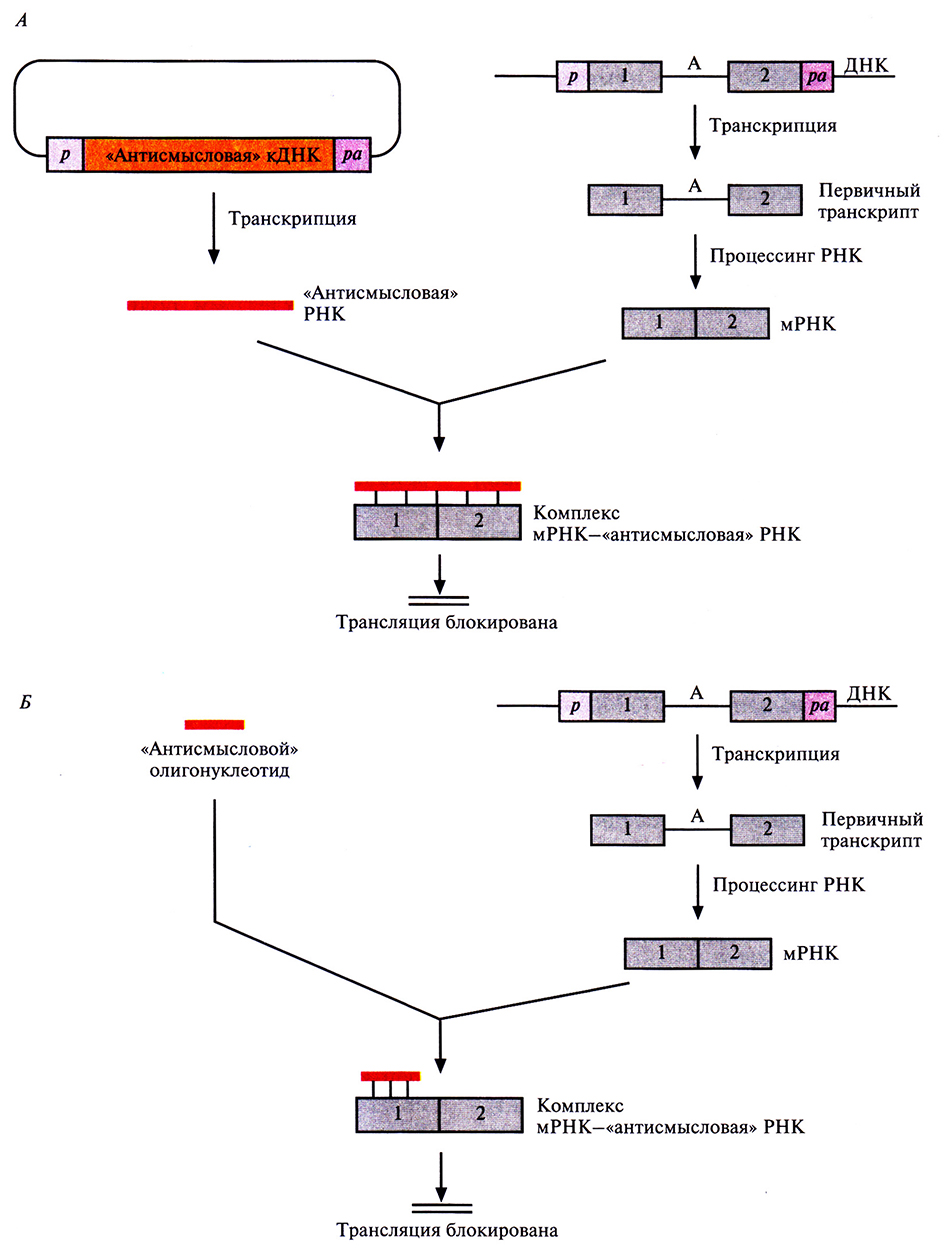
Рис. 21.13. Ингибирование трансляции специфических мРНК с помощью «антисмысловых» нуклеиновых кислот. А. кДНК встраивают в экспрессирующий вектор в обратной ориентации и полученную генетическую конструкцию вводят в клетки, где происходит синтез «антисмысловой» РНК. Эта РНК гибридизуется с мРНК-мишенью и блокирует трансляцию. Б. В клетку вводят антисмысловой олигонуклеотид, который гибридизуется с мРНК-мишенью и блокирует трансляцию. Обозначения: р – промотор, ра – сигнал полиаденилирования, А – интрон, 1 и 2 – экзоны.
Вектор, обусловливающий синтез «антисмысловой» мРНК IGF-1, трансфицировали в культуру клеток глиомы. Если ZnS04 в культуральной среде отсутствовал, то гиперпродукция IGF-1 опухолью сохранялась, если же в среду добавляли ZnS04, то она исчезала. В другом эксперименте изучали последствия введения крысам нетрансфицированных клеток глиомы и клеток, трансфицированных «антисмысловой» кДНК IGF-1. В первом случае опухоли развивались, а во втором – нет.
Если клетки карциномы предстательной железы крыс, трансфицированные «антисмысловой» кДНК IGF-1R, вводили мышам, то у них образовывались лишь небольшие опухоли или не образовывались совсем. Если же им вводили нетрансфицированные или трансфицированные нормальной кДНК IGF-1R клетки, то развивались опухоли большого размера. По-видимому, в обоих случаях «антисмысловая» РНК гибридизуется с комплементарной ей мРНК и ингибирует трансляцию IGF-1 и IGF-1R, предотвращая пролиферацию опухолевых клеток.
«Антисмысловые» олигонуклеотиды как лекарственные средства
Терапевтический эффект синтетических «антисмысловых» олигонуклеотидов зависит от специфичности их гибридизации с доступным сайтом мРНК-мишени, устойчивости к действию клеточных нуклеаз и наличия системы доставки в клетку. 15–20-нуклеотидные последовательности гибридизуются с уникальными мРНК с достаточно высокой специфичностью. Потенциальные сайты-мишени определяют тестированием набора «антисмысловых» олигонуклеотидов с использованием культуры клеток, синтезирующих мРНК-мишень. Для этого проводят электрофоретическое разделение клеточных белков, в которые включают радиоактивную метку во время трансляции, и с помощью радиоавтографии устанавливают, в присутствии какого из «антисмысловых» олигонуклеотидов снижается синтез определенного белка. Никаких общих критериев выбора наилучших сайтов-мишеней в разных РНК-транскриптах не существует. Эффективными могут оказаться олигонуклеотиды, комплементарные 5'- или З'-концам мРНК, границам экзонов и интронов и даже двухцепочечным областям. Олигодезоксинуклеотиды разрушаются внутриклеточными нуклеазами, поэтому важно защитить их от действия последних так, чтобы они не утратили способности к гибридизации с мишенью. Для этого можно модифицировать определенным образом пиримидиновые основания и дезоксирибозу (рис. 21.14). Так, у наиболее широко применяющихся сейчас «антисмысловых» олигонуклеотидов свободный атом кислорода фосфодиэфирной связи заменен на сульфогруппу (рис. 21.14, Б), в результате чего образуется тиофосфатная связь. Модифицированные таким образом олигонуклеотиды растворяются в воде, несут отрицательный заряд и не расщепляются под действием эндонуклеаз. При гибридизации с сайтом-мишенью они образуют РНК–ДНК-дуплексы, которые активируют рибонуклеазу (РНКазу) Н, эндогенный фермент, расщепляющий мРНК в гибридной молекуле. Проведены первые клинические испытания таких олигонуклеотидов – лекарственных средств «первого поколения». Мишенями являются РНК цитомегаловируса, вируса иммунодефицита человека, а также мРНК генов, ответственных за развитие рака, болезней кишечника и других заболеваний.
Синтезированы «антисмысловые» олигонуклеотиды с фосфорамидитной и полиамидной (пептидной) связями (рис. 21.14, В и Г). Такие молекулы очень устойчивы к действию нуклеаз. Химические группы, присоединенные к 2'-углеродному атому сахарного остатка и C-5-атому пиримидинов, также защищают «антисмысловые» олигонуклеотиды и облегчают их связывание с сайтом-мишенью (рис. 1.14, Д и Е). Все преимущества этих и других модификаций сейчас интенсивно изучаются.
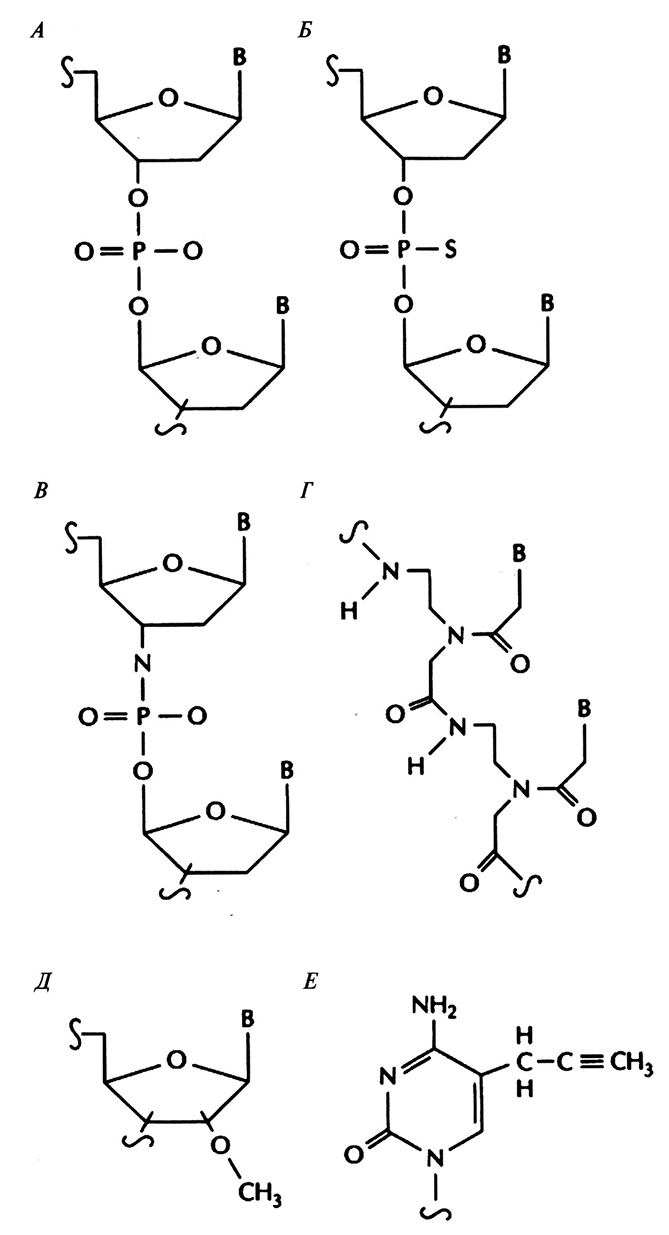
Рис. 21.14. Модификации «антисмысловых» олигонуклеотидов. А. Фосфодиэфирная связь. Б. Тиофосфатная связь. В. Фосфорамидитная связь. Г. Полиамидная связь (пептидная нуклеиновая кислота). Д. 2'-0-метилрибоза. Е. C-5-пропинилцитозин.
Проникновение «антисмыловых» олигонуклеотидов в клетку можно значительно облегчить, поместив их в липосомы. Такая высокоэффективная система доставки позволяет использовать «антисмысловые» олигонуклеотиды в небольших концентрациях. Если же конъюгировать липосомы с сайтами связывания, специфичными для определенных клеток, то можно будет осуществлять адресную доставку олигонуклеотидов.
Проведенные доклинические испытания показали, что «антисмысловые» олигонуклеотиды являются весьма эффективными лекарственными средствами. Изучена возможность их применения для лечения стеноза коронарных и сонных артерий, который приводит к инфарктам и инсультам. В этих случаях часто прибегают к ангиопластике, расширению артерий с помощью баллонного катетера, но примерно у 40% больных через 6 мес вновь возникают стенозы, поскольку ангиопластика стимулирует пролиферацию гладкомышечных клеток и секрецию межклеточного вещества во внутренний слой артерии в месте ее расширения. В одном из экспериментов в сонные артерии крыс после ангиопластики вводили «антисмысловые» олигонуклеотиды с тиофосфатными связями, комплементарные мРНК, которые кодируют важные для клеточного цикла млекопитающих белки; в результате частота повторных стенозов уменьшилась на 90%. Пролиферация гладкомышечных клеток происходит также при атеросклерозе, сахарном диабете, ослсжнениях после коронарного шунтирования. Вероятно, все эти состояния можно будет контролировать аналогичными способами.
«Антисмыловые» олигонуклеотиды можно применять и для лечения вирусных инфекций и малярии. Кроме того, результаты I фазы клинических испытаний лечения болезни Крона с помощью орального введения «антисмыслового» олигонуклеотида проиллюстрировали четко выраженный терапевтический эффект без заметных побочных эффектов. В этом случае мРНК-мишень кодировала межклеточный адгезин типа 1, который вырабатывается в избытке у пациентов с болезнью Крона. Предполагается исследовать эффективность этого же олигонуклеотида для терапии других воспалительных заболеваний, например ревматоидного артрита, псориаза и язвенного колита.
В принципе «антисмысловые» олигонуклеотиды могут образовывать тройную спираль с хромосомной ДНК-мишенью и блокировать транскрипцию. Однако пока специфичность «антигенных» олигонуклеотидов не соответствует стандартам, принятым для лекарственных средств.
Олигонуклеотиды, связывающиеся с белками:
антитромбиновый аптамер
Блокировать экспрессию гена-мишени можно не только с помощью «антисмысловой» терапии, но и введением в клетку олигонуклеотида, связывающегося с фактором транскрипции или трансляции, однако этот подход пока недостаточно изучен. Далее, поскольку нуклеиновые кислоты способны связываться с белками, можно синтезировать такой олигонуклеотид (так называемый аптамер), который будет присоединяться к определенному белку, в норме не связанному ни с какими нуклеиновыми кислотами, и блокировать его функцию. Так, антитромбиновый аптамер может стать недорогим средством профилактики тромбообразования при различных хирургических вмешательствах.
Для его получения использовали набор химически синтезированных олигонуклеотидов, состоящих из 18-нуклеотидных фланкирующих областей (праймеров) и центрального 60-нуклеотидного участка, где в каждом из 60 положений может находиться любой из четырех нуклеотидов. Теоретически такой набор содержит примерно 1,3-1036 (460) олигонуклеотидов с разной центральной последовательностью. Образец пропустили через колонку, содержащую связанные молекулы тромбина, присоединившиеся к тромбину олигонуклеотиды элюировали и повторно пропустили через колонку со связанным тромбином. Эту процедуру повторяли не менее трех раз. Конечный набор тромбиновых аптамеров амплифицировали с помощью ПЦР и клонировали, после чего определили физические и биологические свойства каждого из них. Те аптамеры, которые обладали высокими сродством, специфичностью и антитромбиновой активностью, отбирали для более детального анализа.
Таким образом был получен эффективный антитромбиновый аптамер. К сожалению, вследствие малого времени жизни in vivo его можно использовать только для временного ингибирования функции тромбина (например, при кардиопульмонарном шунтировании), а в тех случаях, когда необходимо длительное введение противосвертывающих веществ (например, при ангиопластике), он неприменим. Очевидно, что описанную процедуру идентификации аптамеров можно использовать и в случае других белков-мишеней.
Рибозимы как лекарственные средства
Рибозимы – это природные РНК, обладающие каталитической активностью (РНК-ферменты); их субстратсвязывающий домен присоединяется к комплементарной РНК-мишени с помощью водородных и, возможно, других связей, а каталитический расщепляет ее в специфическом сайте. Модифицируя субстратсвязывающую последовательность, можно получить рибозим, специфичный в отношении определенной мРНК (рис. 21.15).
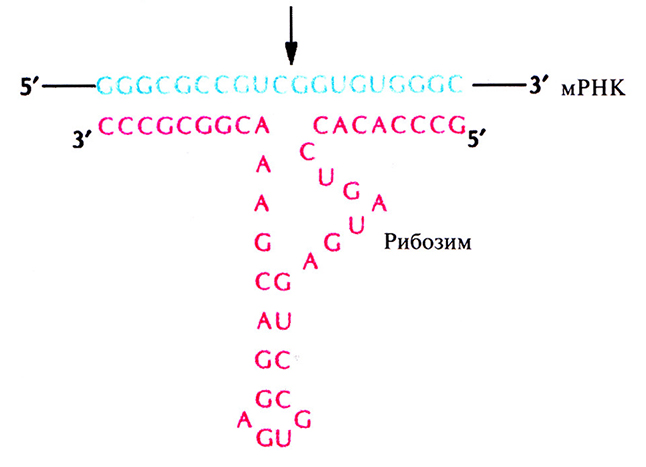
Рис. 21.15. Расщепление мРНК под действием рибозима. Рибозим, субстратсвязывающий домен которого модифицирован с помощью генной инженерии, гибридизуется с мРНК-мишенью и расщепляет ее в специфическом сайте (показано стрелкой). (Из работы Tone et al., In Vivo 7: 471–476, 1993, с изменениями.)
Создание «терапевтического» рибозима – сложный процесс. Связано это с трудностью получения больших количеств синтетических РНК и сохранения их в нативном состоянии в клетке-мишени. В одном из экспериментов синтезировали олигодезоксинуклеотид, который содержал каталитический домен (примерно 20 нуклеотидов), фланкированный гибридизующимися с мРНК-мишенью последовательностями (они же выступали в роли праймеров), амплифицировали его, встроили в эукариотический экспрессирующий вектор и трансфицировали полученной конструкцией клетки. Образовавшийся после транскрипции рибозим расщеплял мРНК-мишень и подавлял трансляцию белка, ответственного за развитие того или иного заболевания. Рибозимы, созданные методами генной инженерии, можно использовать для лечения рака и вирусных инфекций.
Природных ДНК-ферментов (дезоксирибозимов) пока не обнаружено, но уже синтезированы олигодезоксинуклеотиды, обладающие каталитической активностью. Преимущество дезоксирибозимов состоит в том, что для их получения не нужно использовать экспрессирующий вектор: ДНК-ферменты можно упаковать в липосомы и доставить в клетку-мишень. Однако создание эффективных ДНК-ферментов находится пока на начальном этапе развития.
КОРРЕКЦИЯ ГЕНЕТИЧЕСКИХ ДЕФЕКТОВ
С ПОМОЩЬЮ ОЛИГОНУКЛЕОТИДОВ
Многие генетические дефекты можно скорректировать, заменив связанную с данным дефектом пару нуклеотидов в мутантном гене на «правильную» пару. В одном из экспериментов для этой цели использовался 68-членный химерный (ДНК-РНК) олигонуклеотид, который образует шпильку с двумя головками и содержит метилированный кислород при 2'-углеродном атоме рибозы (рис. 21.16). Выбор такого необычного олигонуклеотида основывается на следующих экспериментальных данных: 1) гетеродуплексы РНК-ДНК легче, чем двухцепочечные ДНК, спариваются с гомологичными нуклеиновыми последовательностями; 2) головки шпилек, не участвующие в спаривании, защищают олигонуклеотид от экзонуклеаз; 3) 2'-0-метилирование предотвращает разрушение молекулы РНКазой Н. Важно и расположение нуклеотидов в химерной молекуле: десять рибонуклеотидов фланкируют пять центральных дезоксирибонуклеотидов, причем этот сегмент имеет одинаковую с мишенью последовательность и содержит нормальную пару нуклеотидов.
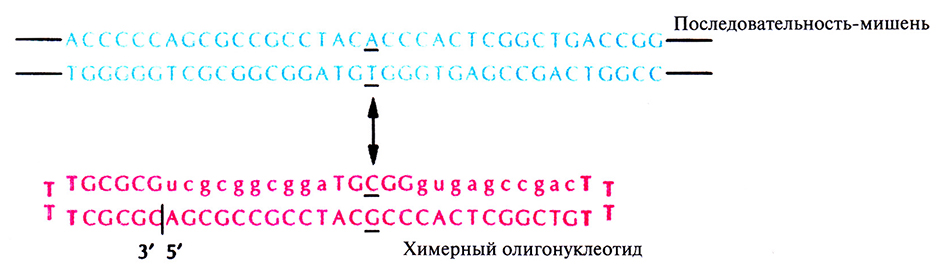
Рис. 21.16. Исправление генетического дефекта, состоящего в замене одной пары нуклеотидов, с помощью химерного олигонуклеотида. Стрелкой указаны мутантный сайт в последовательности-мишени и нормальная пара оснований в химерном олигонуклеотиде. Соответствующие нуклеотиды подчеркнуты. Прописными буквами обозначены дезоксирибонуклеотиды, а строчными – рибонуклеотиды. Жирным шрифтом выделены нуклеотиды, образующие головки шпильки. Вертикальная черта указывает 3'- и 5'-концы химерного олигонуклеотида. (Из работы Yoon et al., Proc. Natl. Acad. Sci. USA 93: 2071-2076, 1996, с изменениями.)
Возможность коррекции мутаций с помощью химерных олигонуклеотидов изучали с использованием как кДНК, входящей в состав плазмиды, так и хромосомной ДНК. В обоих случаях мутантный сайт с высокой частотой заменялся нормальным. Но для того чтобы химерные олигонуклеотиды стали эффективными лекарственными средствами, необходимы дополнительные исследования.
Если мутация в интроне распознается системой процессинга РНК как аутентичный сайт сплайсинга, то в процессированную мРНК включается часть интрона (рис. 21.17, А). Это приводит к сдвигу рамки считывания и образованию укороченного белка. При этом количество нормального белка снижается, что может стать причиной заболевания. Разумно предположить, что если «антисмысловой» олигонуклеотид, комплементарный мутантному интрону, гибридизуется с ним, то ошибочный сплайсинг блокируется, что повысит вероятность сплайсинга в нормальном сайте. Это предположение проверили на β-глобиновом гене с мутацией во втором интроне (рис. 21.17, Б), обусловливающей одну из форм β-талассемии, наследственного заболевания крови, которое приводит к разрушению эритроцитов (анемии). В клетки, гомозиготные по мутантному гену (IVS2-654), ввели содержащий тиофосфатные связи «антисмысловой» 2'-0-метилолигонуклеотид, комплементарный мутантному сайту сплайсинга. В результате число нормальных β-глобиновых цепей увеличилось на 50%. Дальнейшие исследования покажут, является ли этот подход достаточно эффективным для лечения талассемии и других состояний, вызванных подобными мутациями.
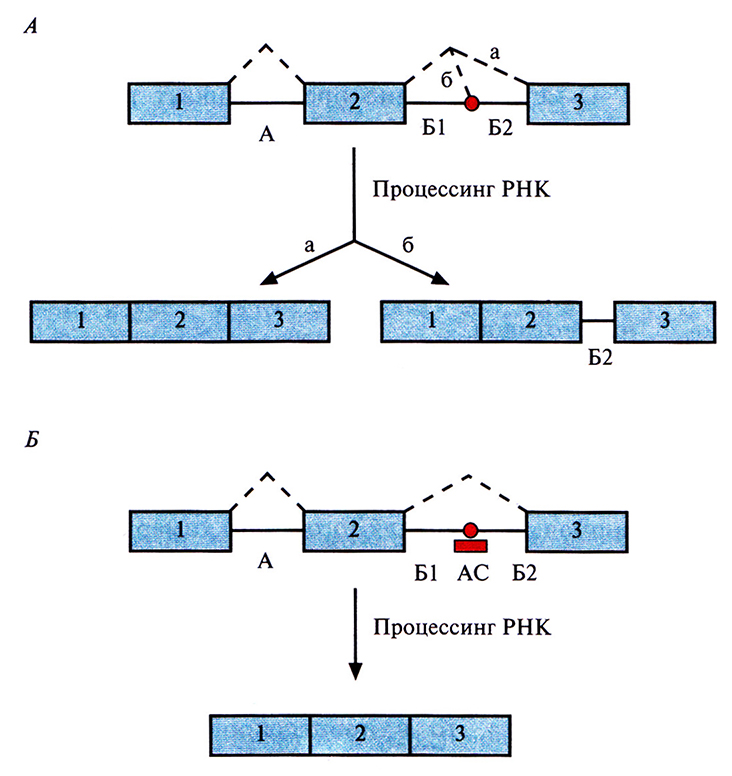
Рис. 21.17. Коррекция дефекта, приводящего к ошибочному сплайсингу, с помошью «антисмыслового» олигонуклеотида. А. Результат мутации, приводящей к ошибочному сплайсингу. Обозначения: цифры – экзоны А – первый интрон, Б – второй интрон, содержащий мутацию (красный кружок), разделяющую его на две части (Б 1 и Б2). Штриховые линии охватывают сегменты РНК, вырезаемые при процессинге. Возможны два варианта сплайсинга: вариант а приводит к образованию функциональной мРНК, вариант б – к образованию РНК, включающей часть второго интрона (Б2). Б. «Антисмысловой» олигонуклеотид (АС), связывающийся с мутантным сайтом сплайсинга, препятствует его распознаванию при процессинге, и в результате образуется только функциональная мРНК.
ЗАКЛЮЧЕНИЕ
Для многих наследственных заболеваний никаких достаточно эффективных способов лечения не существует, и во многом это связано с трудностями получения и адресной доставки соответствующего генного продукта. После разработки методов идентификации и клонирования нормальных вариантов дефектных генов (часто в виде кДНК) были предприняты попытки использовать их для коррекции генетических дефектов. Для лечения заболеваний на молекулярном уровне применяются два основных подхода: генная терапия ex vivo и генная терапия in vivo.
При генной терапии ex vivo «терапевтический» ген переносят в изолированные клетки больного с помощью ретровирусных векторов или других систем доставки, трансдуцированные клетки культивируют и вводят пациенту. При этом у реципиента не развивается нежелательного иммунного ответа, но сама процедура является весьма дорогостоящей и трудоемкой. Альтернативный способ генной терапии ех vivo использует генной нженерную модификацию неаутологичных клеток, заключенных в мембрану, которая предотвращает развитие нежелательного ответа и не препятствует высвобождению «терапевтического» генного продукта.При генной терапии in vivo «терапевтический» ген вводят непосредственно в клетки ткани-мишени больного. Для этого разработаны разные системы доставки: вирусные (ретровирусные, аденовирусные, аденоассоциированные векторы и векторы на основе вируса простого герпеса) и невирусные (инъекция чистой ДНК, бомбардировка ткани-мишени частицами, конъюгированными с ДНК, захват клетками ДНК, заключенной в липидную оболочку). Кроме того, разработань вирусные и невирусные системы доставки генов, специфичные для определенных клеток.
Весьма перспективным способом разрушения быстроделящихся раковых клеток представляется генная активация лекарственного вещества. При этом наиболее широко используется подход, включающий последовательное введение в опухолевые клетки гена тимидинкиназы вируса простого герпеса (HSVtk) и ганцикловира. В результате образуется ганцикловиртрифосфат, токсичный для быстроделящихся клеток. Погибают и трансформированные клетки, контактирующие с HSVtk-модифицированными клетками («эффект свидетеля»).
В качестве лекарственных средств можно использовать не только генные продукты, но и олигонуклеотиды. С помощью так называемых «антисмысловых» олигонуклеотидов можно подавить полностью или частично экспрессию гена того или иного наследственного заболевания. В одном из вариантов такой терапии клонированный ген встраивают в экспрессирующий вектор в обратной ориентации, в результате чего образуется комплементарный нормальной мРНК ДНК-транскрипт, который спаривается с ней и ингибирует тра тсляцию. В более распространенном варианте введенный в клетку-мишень «антисмысловой» олигонуклеотид гибридизуется со специфической мРНК и блокирует ее трансляцию. Эффективность «антисмысловых» олигонуклеотидов и время их жизни повышаются в результате модификаций, затрагивающих фосфодиэфирную связь, пиримидины и остатки Сахаров. В настоящее время изучается терапевтическое действие различных олигонуклеотидов: модифицированных с помощью генной инженерии рибозимов, расщепляющих специфические мРНК; аптамеров, которые связываются со специфическими белками и блокируют их функции; олигонуклеотидов, с помощью которых можно корректировать замену одной нуслеотидной пары и мутации, лриводящие к неправильному сплайсингу.
