- 8.4.1. Пренатальная диагностика наследственных болезней
Многие методы молекулярной генетики начинают широко применяться в пренатальной диагностике наследственных болезней, например гемоглобинопатий. Так, в 1978 г. Кен и Доузи разработали метод диагностики серповидноклеточной анемии путем анализа ДНК из клеток околоплодной жидкости. Это несравненно более безопасный метод, чем взятие для анализа крови- плода, когда вероятность аборта доходит до 7%. Серповидноклеточная анемия — это одно из наиболее часто встречающихся нарушений синтеза гемоглобина. Она развивается в результате замены глутаминовой кислоты в 6-м положении 3-Цепи гемоглобина на валин. При дезоксигенации эритроциты, содержащие- аномальный HbS (две обычные а- и две аномальные 3-цепи), приобретают форму полумесяца (серповидную). Такие негибкие серповидные клетки затрудняют кровообращение в капиллярах и быстро разрушаются. Клиническими проявлениями болезни являются анемия, прекращение роста, приступы острой боли (кризы), а также острые или хронические заболевания органов вследствие закупорки сосудов (например, инсульт).
В работе Кена и Доузи (рис. 8.6) ДНК выделяли из лейкоцитов крови двух родителей, их ребенка, страдающего серповидноклеточной анемией, и здорового добровольца (контроль).
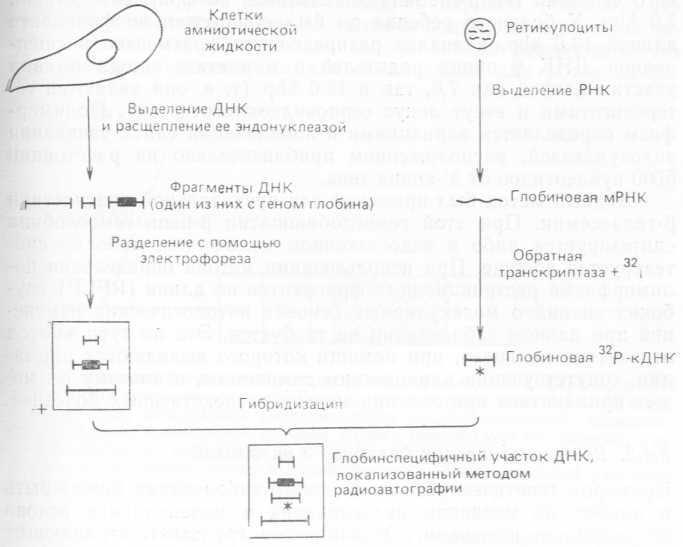
Рис. 8.6. Схема, иллюстрирующая использование генных зондов и анализ полиморфизма длины рестрикционных фрагментов в пренатальной диагностике гемоглобинопатий, например серповидноклеточной анемии (Emery, 1981).
ДНК была выделена также из клеток околоплодной жидкости, отобранной в ходе третьей беременности матери. Вошедший недавно в практику метод картирования с помощью эндонуклеаз рестрикции (гл. 7) позволяет сопоставлять разные ДНК; меченую Р32-кДНК, комплементарную глобиновому гену, можно получить путем выделения глобиновой мРНК из ретикулоцитов и использования обратной транскриптазы, с помощью которой из Р32-нуклеотидов синтезируется кДНК. При расщеплении со- шоставляемых ДНК эндонуклеазой (в данном случае—HpaI) образуются фрагменты разной длины, которые разделяют методом гель-электрофореза. Затем фрагменты переносят на нитроцеллюлозный фильтр и проводят гибридизацию с радиоактивной кДНК-зондом (метод саузерн-блотинг). Нуклеотидные последовательности, связавшие зонд, локализуют методом радиоавтографии. Именно они содержат гены глобина. У здорового человека ген р-глобина локализован во фрагменте длиной 7,6 kbp. У больного ребенка он был обнаружен во фрагменте длиной 13,0 kbp, а анализ распределения фрагментов расщепленной ДНК у обоих родителей и в клетках плода выявил участки длиной как 7,6, так и 13,0 kbp (т. е. они являются гетерозиготами и несут локус серповидноклеточности). Полиморфизм определяется вариациями в локализации сайта узнавания эндонуклеазой, расположенном приблизительно на расстоянии 5000 нуклеотидов от З'-конца гена.
Сходный метод был применен для пренатальной диагностики Р-талассемии. При этой гемоглобинопатии p-цепь гемоглобина синтезируется либо в недостаточном количестве, либо не синтезируется вообще. При использовании метода определения полиморфизма рестрикционных фрагментов по длине (RFLP) глубоких знаний о молекулярных основах патологических изменений при данном заболевании не требуется. Это по сути «метод, отпечатков пальцев», при помощи которого выявляются аномалии, сопутствующие клиническим симптомам, и поэтому он может применяться при лечении многих наследственных болезне..
- 8.4.2. Роль генетических факторов в патологии
Примером генетически обусловленного заболевания может быть, и диабет, но механизм наследования и молекулярная основа его остаются неясными. У пациентов группы 1, страдающих юношеским диабетом, наблюдается полная или почти полная, гибель р-клеток островков Лангерганса, и инсулин у них не образуется. Такая разновидность диабета чаще всего встречается; у гаплотипов Dr3 и Dr4 HLA. В группе 2 (диабет взрослых) уровень инсулина в крови больных близок к норме или повышен; аномалии у них иные, и среди них — нечувствительность, рецепторов к инсулину. Они и приводят к недостатку инсулина. У больных диабетом группы 2 взаимосвязи с определенными типами HLA не выявлено. Ротвейн и др. (Rotwein et al., 1981) использовали метод RFLP для анализа ДНК 35 здоровых людей, 17 больных диабетом из группы 1 и 35 — из группы 2.. У 26% здоровых людей в последовательности ДНК, прилегающей к б'-концу гена инсулина, были обнаружены вставки длиной 1,5—3,4 kbp. Такие же вставки присутствуют и в ДНК 35% больных группы 1 и 66%—группы 2 (рис. 8.7). Была определена полная нуклеотидная последовательность этого полиморфного участка, прилегающего к гену инсулина, и показано, что различия определяются числом и расположением повторяющихся последовательностей. Из ряда опытов следует, что последовательность прилежащего к 5'-концу гена участка может сказываться на экспрессии гена. В этой связи существенно более высокий полиморфизм этого участка при диабете типа 2 представляет особый интерес (следует, впрочем, отметить, что некоторым исследователям не удалось подтвердить наличия такой взаимосвязи). Сегодня мы еще не можем утверждать, что эти изменения достаточны или обязательны для развития болезни, и трактовку результатов нельзя считать однозначной.
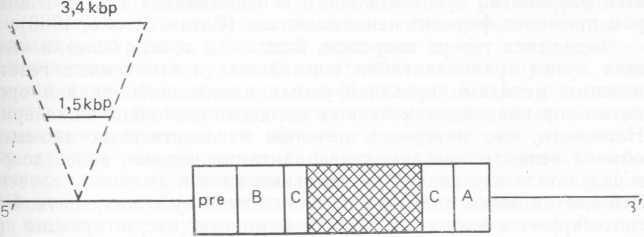
Рис. 8.7. Полиморфизм длины рестрикционных фрагментов 5'-фланкирующего участка гена инсулина человека. Указано место вставок длиной 1,5 и 3,4 kbp. Pre — участок, кодирующий аминокислотную последовательность, примыкающую к N-концу проинсулина, которая, видимо, способствует его секреции в эндоплазматический ретикулум; В, C и А — последовательности, кодирующие В-цепь, конвекторный пептид и A-цепь инсулина. Заштрихованный участок — интрон (гл. 7).
Еще два наследственных заболевания, точная генетическая основа которых нам не известна, но для которых недавно была установлена взаимосвязь между особым типом полиморфизма ДНК и наличием «больного» гена — это мышечная дистрофия Дюшена и хорея Хантингтона. Последнее заболевание неизлечимо, наследуется по аутосомному доминантному механизму и выражается в прогрессирующем слабоумии и параличе, наступающем на тридцатом — сороковом году жизни. К сожалению, до недавнего времени мы не располагали методом выявления носителей такого гена. Поскольку здесь налицо определенная связь с маркерами ДНК, ее следует иметь в виду при генетическом консультировании, и возможно, что со временем на этой основе будут идентифицированы и сам ген хореи Хантингтона, и соответствующий продукт.
