По А.А. Баеву, генетическая инженерия – это конструирование in vitro функционально активных генетических структур (рекомбинантных ДНК), или иначе – создание искусственных генетических программ. Рекомбинантые ДНК придают организму новые генетические и другие свойства.
Возникновение генетической инженерии связано прежде всего с развитием молекулярной биологии. Исследования, проведенные в этой области за последние десятилетия, позволили перейти от описания структуры и функции клеток на уровне органелл к установлению молекулярных механизмов протекающих в них процессов.
В конце 60-х годов многие исследователи считали, что молекулярные механизмы фундаментальных генетических процессов – репликации, транскрипции, трансляции, а также система их регуляции в основном изучены. Стройное, логичное здание молекулярной биологии, казалось, было в основном построено. Схема, показывающая строго однонаправленный поток генетической информации, ДНК → РНК → белок, была названа основной догмой молекулярной биологии и свидетельствовала о незыблемости возведенного здания. Правда, основные положения этой схемы были в основном показаны для кишечной палочки Escherichia coli, которая является прокариотическим организмом, не имеющим истинного ядра. Однако логичность и простота схемы давала возможность считать ее универсальной для всего живого, в том числе и для эукариотических организмов, имеющих ядро, к которым относятся животные, растения и человек.
Однако, несмотря на несомненные успехи молекулярной биологии прокариот, геном сложных организмов был практически недоступен для анализа. Изучение общих биохимических свойств клетки не давало надежды на установление деталей генетической организации: слишком велики были геномы эукариотических организмов и слишком сложно было проводить с ними какие-либо эксперименты. Для этого необходимо было, как минимум, научиться «разрезать» ДНК не в случайных, а в строго определенных местах, с точностью до одного нуклеотида. Возникла и другая проблема – невозможность определения последовательности нуклеотидов в ДНК. Не было выделено ни одного гена, не была расшифрована структура гена. Одна из причин такой ситуации заключается в том, что даже простейшие организмы содержат очень длинные молекулы ДНК (геном кишечной палочки составляет 4,2 • 106 н. п.), а геном высшихэукариот, в том числе растений и млекопитающих, содержит 109–1011 н. п. В геноме содержится несколько десятков тысяч генов.
Сейчас генетическая инженерия позволяет вводить в ядерный аппарат реципиента не только отдельные новые гены и регуляторные последовательности, исходно не присущие данному организму, но и получать новые формы организмов путем введения целых хромосом, отдельных органелл или слияния двух клеток.
1.2. ФЕРМЕНТЫ ГЕНЕТИЧЕСКОЙ ИНЖЕНЕРИИ
Ферменты генетической инженерии – это ферменты, позволяющие проводить различные манипуляции с молекулами ДНК: разрезать в определенных местах, соединять различные по происхождению фрагменты, синтезировать новые, не существующие в природе последовательности, и т. д. Рассмотрим основные ферменты генетической инженерии.
ДНК-полимеразы
Одним из наиболее часто используемых в генетической инженерии ферментов является ДНК-полимераза I, выделенная из Е. coli или фага Т4. ДНК-полимераза I обладает способностью удлинять цепь ДНК в направлении 5' → 3' путем присоединения комплементарного нуклеотида. Это свойство ДНК-полимераз используется в генной инженерии для построения второй комплементарной цепи: при добавлении фермента к одноцепочечной ДНК-матрице в присутствии праймера произойдет ее удвоение. Это свойство используется, например, при создании кДНК-библиотек. ДНК-полимераза применяется также для заполнения «бреши» в цепи ДНК, например, при застраивании фрагментов с выступающими 5'-концами. Экзонуклеазная активность ДНК-полимераз используется для введения радиоактивной метки во фрагмент ДНК.
Использование специфических термостабильных ДНК-полимераз – Tth- и Taq-полимероз – выделенных из бактерий, живущих в гейзерах, позволило проводить амплификацию – множественную наработку любого фрагмента ДНК методом полимеразной цепной реакции (ПЦР). Метод ПЦР, в основу которого положена Taq-полимераза, не только упростил некоторые старые методики генной инженерии, но и позволил проводить молекулярное маркирование как отдельных генов, так и целых геномов.
Из некоторых вирусов была выделена специфическая ДНК-полимераза – РНК-зависимая ДНК-полимераза, названная обратной транскриптазой, или ревертазой. Ревертазы могут синтезировать комплементарную цепь ДНК на РНК-Матрице. С помощью ревертаз можно получать кДНК-ДНК-копиимРНК. кДНК позволяют изучать строение генов и идентифицировать полноценные копии этих генов в геноме.
ДНК-лигаза
Осуществляет одну функцию – соединение фрагментов ДНК путем восстановления фосфодиэфирных связей между соседними нуклеотидами. Этот процесс называется лигированием. Наиболее часто для лигирования в генной инженерии используют ДНК-лигазу фага Т4. С помощью лигазы Т4 соединяют любые фрагменты ДНК с любыми концами: «липкими» или «тупыми» (см. ниже). Это один из наиболее часто использующихся ферментов.
Нуклеазы
Это большая группа ферментов, катализирующих реакцию гидролиза молекул нуклеиновых кислот. В результате действия нуклеаз молекула ДНК или РНК распадается на фрагменты или отдельные нуклеотиды. Исходная функция нуклеаз в клетке – деградация ненужных в данный момент жизнедеятельности молекул (например, деградация мРНК после трансляции) и защита от чужеродных молекул нуклеиновых кислот (расщепление фаговой ДНК бактериальными нуклеазами при заражении бактерии фагом).
Нуклеазы по типу их действия можно поделить на группы. Нуклеазы могут действовать только на молекулы ДНК (ДНКазы) или РНК (РНКазы), либо на молекулы и ДНК, и РНК одновременно (нуклеаза золотистой фасоли). Нуклеазы избирательно могут действовать на одноцепочечную (нуклеаза S1), или двуцепочечную (экзонуклеаза III) молекулы ДНК, или на гибридную ДНК – РНК-молекулу (рибонуклеаза Н).
Кроме того, нуклеазы можно разделить на два типа: на экзонуклеазы и эндонуклеазы. Экзонуклеазы обычно гидролизуют молекулы с 5'- или З'-свободных концов, а эндонуклеазы могут расщеплять внутри последовательности фрагмента или кольцевой молекулы ДНК.
Рестриктазы
Отдельную группу, особенно с утилитарной точки зрения ее применения в генной инженерии, представляют специфические эндонуклеазы – рестриктазы.
В основе метода, позволившего непосредственно приступить к манипуляциям с генами, лежит открытие ферментов, названных рестрикционными эндонуклеазами (рестриктазами). Еще в 1953 г. было обнаружено, что ДНК определенного штамма Е. coli, введенная в клетки другого штамма (например, ДНК штамма В в клетки штамма С), не проявляет, как правило, генетической активности, так как быстро расщепляется на фрагменты ДНК специфическими ферментами – рестриктазами. К настоящему времени из разных микроорганизмов выделено более тысячи различных рестриктаз. В генетической инженерии наиболее широко используются около двухсот.
Рестриктазы представляют собой особый класс эндонуклеаз, которые гидролизуют ДНК строго по определенным специфическим последовательностям, называются сайтами рестрикции. Каждая из рестриктаз узнает свой сайт рестрикции и разрезаетДНКлибо внутри последовательности сайта рестрикции, либо в непосредственной близости от него. Таким образом, при действии конкретной рестриктазы одна и та же последовательность ДНК будет всегда образовывать одинаковый набор фрагментов. Обозначение рестриктаз складывается из начальных букв латинского названия вида бактерий, из которого был выделен фермент, и дополнительного обозначения, так как из бактерий одного вида может быть выделено несколько различных рестриктаз: Escherichia coli – EcoR I, EcoR V, Haemophilus influenzae – Hinf I, Sreptomyces albus – Sal I, Thermus aquaticus – Taq I.
Рестриктазы делятся на несколько типов по характеру расщепления нуклеотидной последовательности. Рестриктазы I типа узнают сайт рестрикции, но расщепляют последовательность ДНК на произвольном расстоянии (от нескольких десятков до нескольких тысяч пар нуклеотидов) от сайта узнавания. Такие рестриктазы невозможно использовать для решения генно-инженерных задач. Рестриктазы III типа похожи на рестриктазы 1 типа, они гидролизуют ДНК на расстоянии 20–35 н. п. от сайтов узнавания и также довольно редко используются для практических целей.
Ферменты, используемые для получения рекомбинантных молекул,– рестриктазы II типа. Основной характеристикой таких рестриктаз является то, что у них сайты узнавания и места рестрикции совпадают. Обычно рестриктаза II типа узнает определенную последовательность на ДНК и гидролизует ее внутри последовательности сайта рестрикции. Сайты рестрикции рестриктаз II типа представлены симметричными при повороте на 180° последовательностями – палиндромами:
5' GAATTC 3'
У CTTAAG 5' сайт рестрикции рестриктазы EcoR I
5' TAGA 3'
3' АТСТ 5' сайт рестрикции рестриктазы Taq I
Рестриктазы II типа делятся на несколько классов в зависимости от размера сайта рестрикции и длины получаемых фрагментов ДНК:
1) мелкощепящие – сайт рестрикции которых представлен четырьмя нуклеотидными парами;
2) среднещепящие – сайт рестрикции – 6–8 н. п.;
3) крупнощепящие – сайт рестрикции – 10–14 н. п.
Рестриктазы II типа можно отнести к двум группам по тому, как они расщепляют последовательность ДНК. Одни вносят разрывы по оси симметрии узнаваемой последовательности, а другие – со сдвигом, с образованием «ступеньки». В первом случае образуются так называемые «тупые» концы, а во втором – «липкие», т. е. фрагменты имеют на своих концах однонитевые взаимно комплементарные участки.
Образование при расщеплении рестриктазами фрагментов с «липкими» концами:
5' G ↓ ААТТС З' 5' С ↓ CGG З'
EcoR I 3 СТТАА ↑ G 5 Нра II З' GGC ↑ С 5'
Образование при расщеплении рестриктазами фрагментов с «тупыми» концами:
ТА ↓ I GA З' 5' GTT ↓ ААС З'
Taq I З' AT ↑ СТ 5' Hinc I З' САА ↑ TTG 5'
Фрагменты ДНК, имеющие одинаковые «липкие» концы, могут соединяться друг с другом с помощью ДНК-л игазы, при этом сайт рестрикции восстанавливается. Фрагменты, имеющие «тупые» концы, могут быть соединены вне зависимости оттого, какой рестриктазой они были образованы. Фрагменты с «липкими» концами более удобны для создания рекомбинантных ДНК, так как ДНК-лигаза обеспечивает беспрепятственное соединение фрагментов. Ферментативная активность рестриктаз измеряется в единицах активности. Это такое количество фермента, которое необходимо для полного гидролиза за один час 1 мкг ДНК фага λ при оптимальных условиях. Оптимальные условия рестрикции для каждой рестриктазы являются индивидуальными и зависят от рН, ионной силы, присутствия определенных ионов, температуры проведения реакции. Рестриктазы являются основными ферментами, используемыми в генетической инженерии.
1.3. РАЗДЕЛЕНИЕ ФРАГМЕНТОВ ДНК
И ПОСТРОЕНИЕ РЕСТРИКЦИОННЫХ КАРТ
(ФИЗИЧЕСКОЕ КАРТИРОВАНИЕ)
Ферменты рестрикции стали эффективным инструментом исследования. Они позволяют превращать молекулы ДНК очень большого размера (106–1011 н. п.) в набор фрагментов длиной от нескольких сотен до десятков тысяч пар оснований. С помощью метода электрофореза в агарозном геле фрагменты ДНК, различающиеся по размеру, можно легко разделить, а затем исследовать каждый фрагмент отдельно. Метод электрофореза основан на разделении (фрагментов) молекул ДНК, движущихся с различной скоростью в электрическом поле. В растворе ДНК существует в виде аниона, и при помещении раствора ДНК в электрическое поле молекулы будут двигаться к положительному полюсу (катоду).
Разделение фрагментов ДНК осуществляют в носителе, которым является раствор полимера агарозы. Отсюда и название метода. Агарозный гель образует трехмерную полимерную ячеистую структуру. Он электронейтрален и химически инертен по отношению к ДНК, поэтому всегда легко можно выделить (элюировать) необходимый фрагмент ДНК из геля с сохранением биологической активности. Использование геля в качестве среды, где проводится электрофорез, позволило решить проблему разделения фрагментов и затем выделения конкретного фрагмента ДНК (рис. 1.1).

Рис. 1.1. Разделение фрагментов ДНК, полученных в результате действия рестриктаз с помощью электрофореза в агарозном геле:
а-ДНК фага λ Hind III; ДНК фага X/Нае III; ДНК плазмиды pBR322/Bst N1 (в знаменателе дроби – название рестриктазы);
б – схематичное изображение фрагментов ДНК фага λ, полученных под действием различных рестриктаз (на шкале указаны размеры фрагментов в н. п.)
Короткие фрагменты в агарозном геле мигрируют намного быстрее, чем длинные, при этом подвижность фрагментов ДНК в геле обратно пропорциональна логарифму массы (заряда) этого фрагмента. При окрашивании гелей красителями (например, бромистым этидием), связывающимися с ДНК, выявляется набор полос, каждая из которых отвечает рестрикционному фрагменту. Молекулярную массу фрагмента можно определить, проводя калибровку с помощью фрагментов ДНК с известными молекулярными массами. Использование электрофореза для разделения рестрикционных фрагментов дает возможность получать рестрикционные карты – последовательности ДНК с нанесенными на них сайтами разрезания для различных рестриктаз. Первая карта была получена для вируса SV40, содержащего 5423 пары оснований (рис. 1.2). Использовали рестриктазу Hind III, расщепляющую кольцевую ДНК вируса на 11 фрагментов. Порядок их расположения в ДНК был установлен путем исследований наборов образованных фрагментов. Первый разрыв превращал кольцевую молекулу в линейную, которая затем расщеплялась на все меньшие фрагменты. Вначале исследовали наборы перекрывающихся фрагментов, а затем продукты полного расщепления. Таким образом была получена рестрикционная карта кольцевой вирусной ДНК, на которую были нанесены сайты расщепления рестриктазой Hind III. Повторив подобные эксперименты с другими рестриктами, можно получить более подробную карту, где отмечены сайты рестрикции для нескольких рестриктаз. Чем больше взято рестриктаз для картирования, тем более подробна карта.
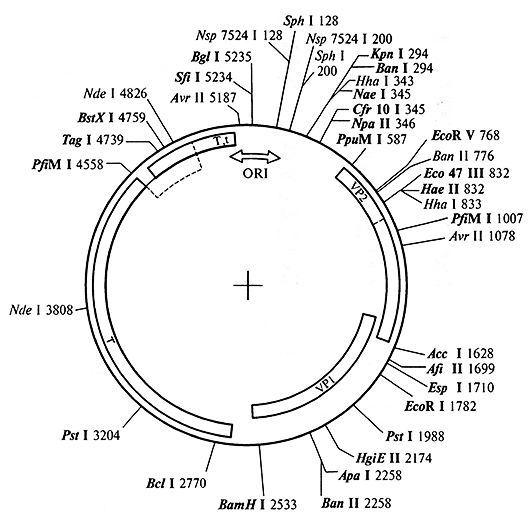
Рис. 1.2. Генетическая и рестрикционная карта ДНК вируса SV40 (цифрами указано положение сайтов рестрикции)
Определение нуклеотидной последовательности – секвенирование
Методы, позволившие идентифицировать генетически важные участки ДНК, имели большое значение. Но они также способствовали разработке исключительно эффективных новых методов секвенирования ДНК и создания рекомбинантных молекул. Секвенирование позволяет довольно быстро определить полную нуклеотидную последовательность сегмента длиной 350– 1000 нуклеотидных пар и более, образующегося при расщеплении ДНК рестрикционными эндонуклеазами. Существует два основных принципа секвенирования: химический и ферментативный сиквенс.
Химический сиквенс основан на избирательной химической деградации нуклеотидов. Он был предложен в 1977 г. A.M. Максамом и В. Гилбертом и назван их именем. Для секвенирования этим методом необходимо получить одноцепочечную молекулу ДНК, один из концов которой метят с помощью изотопа фосфора 32Р; препарат меченой ДНК делят на четыре порции и каждую обрабатывают реагентом, специфически разрушающим одно или два из четырех оснований. Так, например, добавление 60 %-й муравьиной кислоты разрушает пуриновые основания (А + Г), прибавление диметилсульфата – только гуанидиновые основания; чистый гидразин разрушает пиримидиновые основания (Т + Ц), а в присутствии 1,5 М NaCI избирательно разрушаются только цитозиновые основания. Очень важно подобрать условия реакции таким образом, чтобы на каждую молекулу ДНК приходилось лишь несколько повреждений. При обработке поврежденных молекул пиперидином в ДНК образуется разрыв в том месте, где находилось разрушенное основание.
В результате получается набор меченых фрагментов, длины которых определяются расстоянием от разрушенного основания до конца молекулы. Фрагменты, образовавшиеся во всех четырех реакциях, подвергают электрофорезу на четырех соседних дорожках в акриламидном геле в денатурирующих условиях; затем проводят радиоавтографию, и фрагменты, содержащие радиоактивную метку, оставляют «отпечатки» на рентгеновской пленке. По их положению можно определить, на каком расстоянии от меченого конца находилось разрушенное основание, а зная это основание – его положение. Так, по набору полос на рентгеновской пленке определяют нуклеотидную последовательность анализируемого фрагмента ДНК.
С помощью этого метода за короткий срок удалось установить полную нуклеотидную последовательность ДНК SV 40, рекомбинантной плазмиды pBR322 и многих других последовательностей ДНК. Однако секвенирование методом Максама–Гилберта имеет ряд недостатков, связанных с длительностью и значительной трудоемкостью процедур. В настоящее время на основе секвенирования с помощью химической деградации разработаны усовершенствованные и быстрые методы определения нуклеотидной последовательностости, в том числе твердофазное секвенирование и секвенирование с использованием обращенно-фазовой хроматографии.
Наиболее широко сейчас применяется метод ферментативного секвенирования, или метод секвенирования путем терминации (остановки синтеза) цепи, предложенный Ф. Сэнгером в 1977 г. (рис. 1.3, а). В основе метода Сэнгера лежит принцип репликации комплементарной цепи ДНК на одноцепочечной матрице при происходящем в разных местах последовательности ДНК обрыве синтеза, т. е. терминации роста цепи. Основным моментом ферментативного секвенирования является терминация синтеза строящейся цепи. Терминирующими агентами, приводящими к остановке репликации, являются 2', З'-дидезокситрифосфаты (ддАТФ, ддГТФ, ддТТФ, ддЦТФ). Эти модифицированные основания не могут образовать фосфодиэфирную связь со следующим дезоксирибонуклеотидом. В результате рост (элонгация) данной цепи терминируется в том месте, где в ДНК включился дидезоксирибонуклеотид.
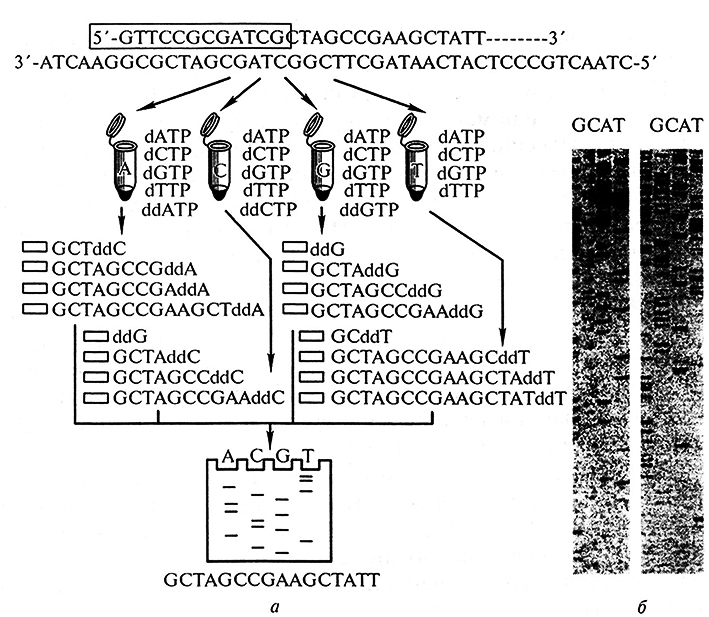
Рис. 1.3. Принцип определения нуклеотидной последовательности ДНК ферментативным методом Сэнгера с дидезокситерминантами (по Чемерис и др., 1999) (а) и радиограмма, получаемая в результате эксперимента (б)
Так как в основе секвенирования по Сэнгеру лежит процесс репликации ДНК, то основным ферментом является ДНК-полимераза (используют ДНК-полимеразу I). В присутствии одноцепочечной ДНК-матрицы, короткогополинуклеотидного праймера и нуклеотидов по принципу комплементарности будет происходить синтез второй цепи ДНК. При этом удлинение цепи будет проходить до тех пор, пока вместо дезоксинуклеотида не присоединится дидезоксинуклеотид. Присоединение последнего приведет к остановке синтеза.
В пробирках собирают четыре реакционные смеси, имеющие одинаковый состав: денатурированный фрагмент ДНК-матрицы, чью нуклеотидную последовательность определяют, четыре дезоксинуклеотида, радиоактивно меченый короткий (16-24 н. п.) праймер, комплементарный фрагменту ДНК-матрицы, с которого ДНК-полимераза I продолжит синтез, и сама ДНК-полимераза I. В каждую пробирку затем добавляют только один издидезоксинуклеотидов. В каждой пробирке синтезируемая цепь будет терминироваться в том месте, где ДНК-полимераза I присоединит дидезоксинуклеотид, который был добавлен в реакционную смесь, т. е. в одной пробирке синтез оборвется при присоединении к комплементарной цепи ддАТФ, в другой – ддГТФ и т. д. Поскольку обрыв цепи происходит в случайных местах, то получается набор фрагментов всех возможных длин, начиная с праймера до конца секвенируемого фрагмента.
Полученные фрагменты ДНК разделяют в полиакриламидном геле (с точностью до одного нуклеотида), проводят радиоавтографию и по картине распределения фрагментов в четырех пробах устанавливают нуклеотидную последовательность ДНК (рис. 1.3, б).
Располагая такой информацией, можно локализовать на ДНК биологически важные участки. Так, например, показано, что репликация вируса SV 40 всегда начинается в одном специфическом Hind ///-фрагменте и продолжается в обоих направлениях. Рестрикционные карты и рестрикционные фрагменты были также использованы для картирования участков ДНК, на которых синтезируются мРНК для вирусных белков. В настоящее время определение точной нуклеотидной последовательности любого фрагмента ДНК – вполне разрешимая задача. Уже определена последовательность нескольких тысяч генов про- и эукариот. В настоящее время расшифрованы последовательности большинства прокариотическихорганизмов. Изэукариот полностьюсеквенированы геномы дрожжей (S. cerevisiae), нематоды (С. elegans),арабидопсиса, дрозофилы и человека. На подходе расшифровка геномов риса и мыши. Понятно, что проведение работ такого масштаба невозможно без автоматизации и модернизации методов секвенирования. Уже полностью налажено автоматическое секвенирование на основе обоих методов, что значительно упрощает и удешевляет секвенирование. Особенно широко используется автоматизированный ферментативный метод определения нуклеотидных последовательностей. Применение флуоресцентных красителей, связанных с терминирующими нуклеотидами, позволяет проводить все реакции в одной пробирке. Кроме того, разработаны принципиально новые подходы к секвенированию, например, секвенирование посредством гибридизации с фиксированными олигонуклеотидными чипами, секвенирование посредством использования экзонуклеаз и ряд других методов. Все эти методы позволяют довольно быстро решать проблемы секвенирования как огромных участков ДНК, так и любых геномов. Полученные результаты заносятся в базы данных – банки генов. Наиболее крупными являются банки генов NCBI (США), EMBL (ОЕ), DDBJ (Япония), объединенных в единую сеть INSD. Нуклеотидные последовательности, представленные в этих банках данных, доступны по адресам в Интернете: www.ncbi.nlm.nih.gov, www.ddbj.nig.ac.jp,www.ebi.ac.uk.
Зная нуклеотидную последовательность гена и генетический код, легко определить аминокислотную последовательность кодируемого им белка. Раньше для определения структуры белка приходилось делать тщательный и весьма трудоемкий анализ выделенного и очищенного белка. Сейчас часто бывает проще определить структуру белка через нуклеотидную последовательность, чем с помощью прямого секвенирования белка. Существует огромное количество компьютерных программ, позволяющих проводить компьютерный анализ полученных секвенированных последовательностей, в том числе сравнение с уже известными последовательностями, определение степени гомологии, выявление специфических (например, регуляторных) последовательностей, возможность моделировать вторичные структуры РНК.
Сразу вслед за разработкой быстрых методов секвенирования появились столь же быстрые и простые методы синтеза сравнительно длинных олигонуклеотидов с определенной, заранее заданной последовательностью. Теперь довольно легко можно синтезировать последовательность до 100 нуклеотидов. Автоматизация этой процедуры еще более облегчает и ускоряет синтез.
